
MA: Marketing Authorization
Who is concerned by the MA?
Medicinal products must receive an MA (L5121-8 Public Health Code) before being introduced onto the European or French market.
A medicinal product is defined (L 5111-1 of the Public Health Code or Council Directive 65/65/EEC of 26 January 1965, Article 1) as:
- a substance or composition presented as having curative or preventive properties with respect to diseases, or
- a substance or composition which may be administered to establish a medical diagnosis, or a substance or composition which may be administered to restore, correct or modify physiological functions.
Main stages in the development of a medicinal product
Generally, these are the main stages in the lifecycle of a medicinal product, from development to marketing.
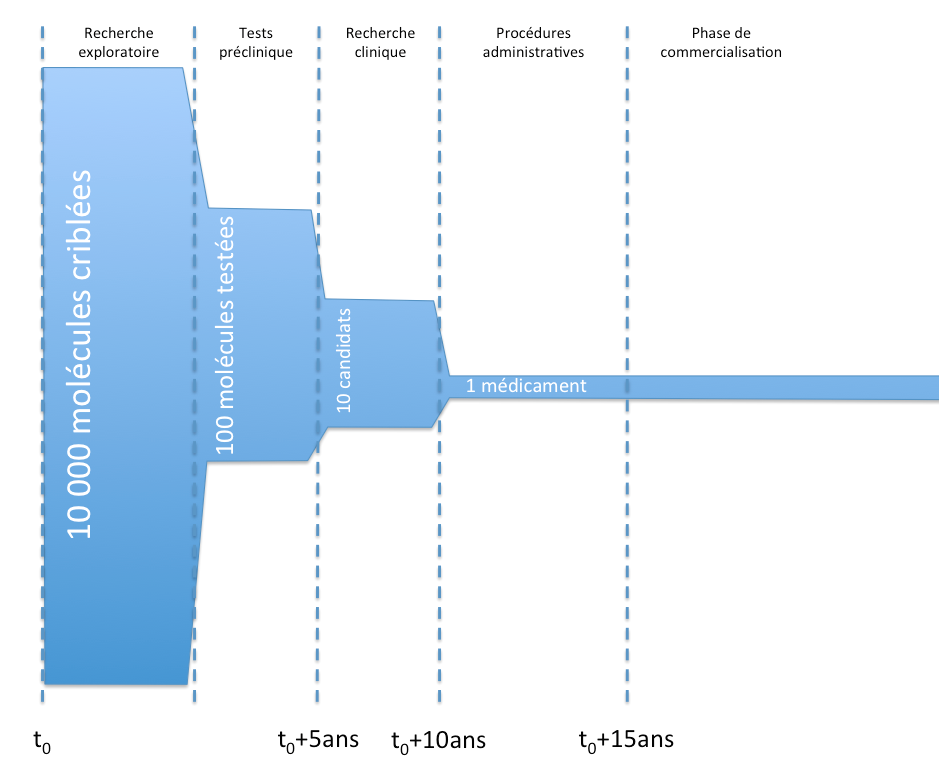 Lifecycle of a medicinal product and MA
Lifecycle of a medicinal product and MAThe purpose of preclinical trials is, in particular, to verify the tolerance of molecules in vitro in animals, to conduct toxicological tests and to study the pharmacokinetics of the molecules (in short, to observe how quickly the molecules act, are absorbed, or are eliminated).
Clinical research is divided into three main phases:
- Phase I:
- trials in human patients, on a small cohort of healthy volunteers.
- These trials aim, in particular, to test the tolerance of the molecules in patients.
- Phase II:
- trials in human patients, on a larger cohort of sick volunteers (but without other pathologies).
- These trials aim, in particular, to establish a benefit/risk ratio for the molecule and to define effective doses.
- Phase III:
- trials in human patients, on a large cohort of sick volunteers under real-life conditions.
- These trials aim, in particular, to verify efficacy (« double-blind » study) and long-term safety.
During the marketing phase, a « pharmacovigilance » system (L5121-22 of the Public Health Code et seq.) is implemented: potential side effects are collected from healthcare professionals and may lead to observational studies.
In addition, interventional studies (known as Phase IV) may be conducted, particularly if the MA requires it due to the small number of patients during clinical trials (L5121-8-1 Public Health Code).
Finally, during the marketing phase, secondary clinical studies may be conducted at the initiative of the manufacturer to test its medicinal product on another pathology, a new population, as part of a new therapeutic strategy, etc., in order to obtain authorization for a new indication.
Procedures and modalities
Different possible procedures
To obtain an MA, different routes may be chosen:
- National procedure:
- is, of course, valid for only one country.
- Centralized procedure: covers 27 European countries; may be mandatory for certain medicinal products (e.g., biotechnologies or new substances in certain fields such as cancer or HIV); mutual recognition procedure: an MA obtained in one reference country may be recognized by other countries; decentralized procedure: similar to the mutual recognition procedure, but no MA has yet been obtained. Applications are filed in all countries but are examined only by one reference country.
File
The filing of a marketing authorization (MA) application is submitted to the competent authorities (French, European, etc.) in a format that is fortunately harmonized (CTD format).
The MA file includes in particular the following information:
- administrative (this depends on the competent authorities);
- on quality (manufacturing process, raw materials used, product stability, etc.);
- on preclinical trials;
- on clinical research;
- on the information to be included on packaging and the patient information leaflet (in the annex).
Decision-Making
On this basis, the competent authorities may grant the MA based on the quality, efficacy, and safety information of the medicinal product: a benefit/risk ratio is then established, taking into account other available treatments (L5121-9 Public Health Code).
The decision is important, as it determines the name, form of the medicinal product, method of administration, dosage, therapeutic indications, prescriber, and duration of treatment… All these elements will indirectly set the price and thus the profitability of the medicinal product.
Protection of MA Data
Principle
The MA file is not public for a certain period, known as the « data protection » period.
During the « data protection » period, the regulatory authorities may not:
- disclose the file, nor
- allow another company to refer to a third party’s MA to obtain an authorization.
The duration of the « data protection » period is 8 + 2 + 1 years (Regulation (EC) No 726/2004, art. 14(11)):
- 8 years of protection is granted, during which no one may access or refer to the MA file (R5121-28 Public Health Code);
- 2 additional years of protection, during which it is possible to refer to the MA file, but during which it is not possible to obtain an MA on this basis (L5121-10-1 Public Health Code);
- 1 additional year of protection (in addition to the previous 2 years), if during the 8-year period a new therapeutic indication is granted and it provides a significant benefit (L5121-10-1 Public Health Code).
End of MA File Protection and Existence of a Patent
If the MA file is accessible and a third party obtains a new MA on this basis (end of the 8+2+1 period), this does not mean that they will not be an infringer if a patent exists and is in force.
It is important to clearly distinguish between these two issues.
Protection and Line Extension
If, after obtaining an MA, a new therapeutic form or a new dosage is authorized (via a secondary clinical study), this does not mean that a new MA is granted.
Thus, a line extension does not allow the extension of the protection period of an MA (R5121-41-1 Public Health Code or Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001, art 6.1).
Protection and Orphan Disease
An orphan disease affects no more than 5 persons per 10,000 in the Community (or the marketing of a medicinal product does not generate sufficient profit to justify the investment) and no medicinal product has yet been authorized (at least, with comparable efficacy) (Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999, Article 3(1)).
If a marketing authorization (MA) is sought for an orphan disease, there is a guarantee that no other MA (including an MA extension) will be granted for a similar medicinal product (Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999, Article 8.1) for 10 years.
This 10-year period may be reduced to 6 years if it is established before the end of the 5th year that the disease no longer qualifies as an orphan disease as defined above (Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999, Article 8.2).
However, this 10-year exclusivity may be reduced if (Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999, Article 8.2):
- the proprietor gives consent;
- the proprietor is unable to produce the medicinal product in sufficient quantity;
- a new applicant demonstrates that their medicinal product is safer, more effective, or clinically superior.
Protection and Pediatric Orphan Disease
The 10-year period mentioned for orphan diseases in the previous section is extended to 12 years if the MA application includes results from all studies conducted under an approved pediatric investigation plan (Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006, Article 37).
It should be noted that if a supplementary protection certificate (SPC) exists, there are specific provisions regarding pediatric studies (see below).
Pricing and Reimbursement
Pricing
The price of the medicinal product is determined through negotiations between manufacturers and the Economic Committee for Health Products (CEPS). If no agreement is reached, the CEPS unilaterally sets the price. The price, established by ministerial order (Article L5123-1 of the Public Health Code), takes into account:
- the prices of other medicinal products with the same therapeutic aim;
- the anticipated sales volumes;
- the conditions of use;
- the improvement over existing treatments (or ASMR, improvement in actual benefit).
Reimbursement
Based on the actual benefit (SMR), a decision is made as to whether the medicinal product is fully or partially covered by social security. Other medicinal products are not considered in this assessment.
The final decision on reimbursement listing falls under the competence of the ministers responsible for Health and Social Security (Article L162-17-2-1 of the Social Security Code).
The SMR may be:
- major: reimbursement at 65% to 100% (the maximum rate is granted for irreplaceable and costly medicinal products);
- moderate: reimbursement at 30%;
- low: reimbursement at 15%;
- insufficient: not reimbursed.
Generics and Biosimilars
Definition
A generic is a medicinal product having the same qualitative and quantitative composition in active substance and the same pharmaceutical form as a reference medicinal product (Article L5121-1 of the Public Health Code, 5° a or Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001, Article 10.2 b).
Additionally, bioequivalence between the generic and the reference medicinal product must be demonstrated.
Simplified MA Procedure
The marketing authorization (MA) application for a generic is simplified.
Thus, the applicant is not required to provide results:
- of pharmacological and toxicological tests;
- of clinical trials.
Only pharmaceutical data corresponding to the quality of raw materials and the manufacturing process are required.
Furthermore, it must be demonstrated that the generic is similar to a medicinal product that has obtained an MA for at least 10 years (Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001, Art 10.1) via a bioequivalence study (i.e., equivalent behavior in the body: absorption, distribution, metabolism, and elimination).
Possible Differences
It is therefore possible to have a different presentation (e.g., syrup vs. pill) or different excipients.
It is also possible to have a different therapeutic indication (i.e., list of pathologies for which the medicinal product may be used, Conseil d’État, 23 July 2003, No. 246716).
Price Determination
The price is determined as for a standard medicinal product, but it will be lower than the reference medicinal product, as research costs are lower.
Supplementary Protection Certificates or SPCs
Principle
SPCs were created to take into account the particularly long duration required to obtain an MA and to prevent the protection from being lost even before the start of the medicinal product’s commercialization.
Applicable Legislation?
The existence of the SPC is provided for by:
- the Council Regulation No 1768/92 of 18 June 1992:
- for SPCs granted after 2 January 1993 (Article 23 of the Regulation) if the patent covers a medicinal product (Article 2 of the Regulation), its process of manufacture, or its use (Article 2 of the Regulation together with Article 1(c));
- the Regulation (EC) No 1610/96 of the European Parliament and of the Council of 23 July 1996:
- for SPCs granted after 8 February 1997 (Article 23 of the Regulation) if the patent covers a plant protection product (Article 2 of the Regulation);
- Articles L611-2 IPC and L611-3 IPC:
- for SPCs granted before 2 January 1993.
Needless to say, the provisions of the IPC are no longer widely applicable today…
Independent Title
An SPC is an independent title; it does not extend the duration of a patent (even though the existence of the patent is a prerequisite): it confers its own protection attached to that SPC.
Conditions for the Grant of SPCs
To obtain an SPC, the following conditions must be met:
- file an application
- hold a patent in force (and not merely a patent application) covering one of the elements mentioned above, depending on the applicable text;
- Article 3(a) of Regulation No 1768/92 of the Council of 18 June 1992 or
- Article 3(a) of Regulation (EC) No 1610/96 of the European Parliament and of the Council of 23 July 1996;
- hold a valid marketing authorization (MA) covering a medicinal product (i.e., the commercial name of a medicinal product) covered by the patent, or if the patented medicinal product or plant protection product has been the subject of an MA
- Article 3(b) of Regulation No 1768/92 of the Council of 18 June 1992 or
- Article 3(b) of Regulation (EC) No 1610/96 of the European Parliament and of the Council of 23 July 1996);
- not have previously filed an application for an SPC (
- Article 3(c) of Regulation No 1768/92 of the Council of 18 June 1992 or
- Article 3(c) of Regulation (EC) No 1610/96 of the European Parliament and of the Council of 23 July 1996);
- ensure that this MA is the first in the Community
- Article 3(d) of Regulation No 1768/92 of the Council of 18 June 1992 or
- Article 3(d) of Regulation (EC) No 1610/96 of the European Parliament and of the Council of 23 July 1996);
Scope of Protection
The protection conferred by the SPC is limited to the medicinal product covered by the MA (L611-3 IPC or Article 4 of Regulation No 1768/92 of the Council of 18 June 1992 or Article 4 of Regulation (EC) No 1610/96 of the European Parliament and of the Council of 23 July 1996).
Furthermore, it is not possible to obtain an SPC that is broader (CJEU C-518/10 of 25 November 2011) or narrower (CJEU C-322/10 of 24 November 2011) than the claim of the referenced patent (exact correspondence): if the claim covers A1+A2, it is not possible to obtain an SPC for A1+A2+A3 (even if the MA is granted for A1+A2+A3) or for A1 alone.
Duration of Protection
Principle
The SPC allows for a protection period not exceeding (Article 13 of Regulation No 1768/92 of the Council of 18 June 1992 or Article 13 of Regulation (EC) No 1610/96 of the European Parliament and of the Council of 23 July 1996):
- 5 years from the expiration of the patent;
- and the time difference between the date of the first MA and the filing date of the patent, from which 5 years are subtracted.
In summary, the duration of protection granted by the SPC is:
- if the MA is granted more than 10 years after the filing of the patent application: 5 years;
- if the MA is granted in less than 10 years, the 5-year duration is reduced accordingly.
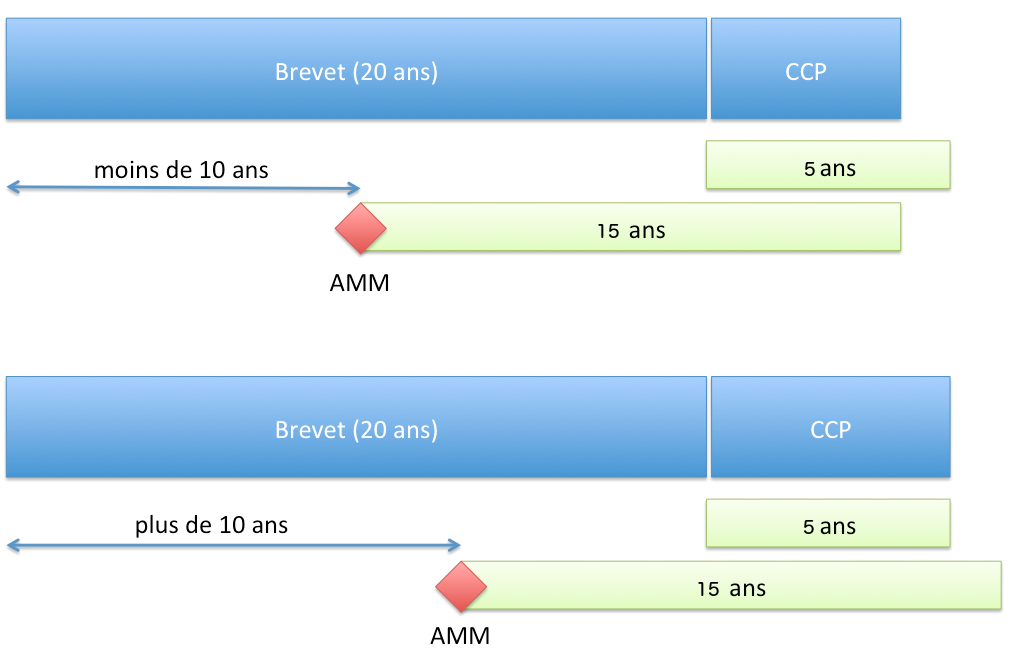 Duration of protection conferred by an SPC (EU Regulation)
Duration of protection conferred by an SPC (EU Regulation)Starting point of the time limit
The question that arose was what the « date of the first marketing authorization » referred to:
- the date of the decision granting the MA or
- the date of notification of that decision or
- the date of grant.
This can be significant…
The CJEU had the opportunity to rule on the matter (CJEU C‑471/14, 6 October 2015) and stated that the « date of the first marketing authorization » was the date of notification of that decision.
Negative duration?
Due to the calculation method mentioned above, it is entirely possible for the duration of an SPC to be negative.
Although the patent proprietor is usually not interested in obtaining an SPC with a negative duration, they may exceptionally wish to do so, particularly if they intend to benefit from a pediatric extension mentioned below.
The CJEU stated that this was entirely possible (CJEU C‑125/10, 8 December 2011).
Pediatric extension
If the proprietor of an SPC provides the results of studies conducted under an approved pediatric investigation plan (in whole or in part for the 0 to 18 age group) in their MA or supplementary MA filing, they are entitled to a 6-month extension of their SPC (Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006, Art 36.1).
However, this extension is not possible:
- if the medicinal product is designated in the MA as targeting a pediatric orphan disease (additional MA protection is already granted, see above) (Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006, Art 36.4);
- if the MA benefits from an additional one-year protection granted in the case of a new therapeutic indication (see above) (Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006, Art 36.5).
Dans quel délai peut-on demander le CCP ? merci d’avance