
Das Zulassungsverfahren: « Marktzulassung »
Wer ist von der Marktzulassung betroffen?
Arzneimittel müssen vor ihrer Einführung auf dem europäischen oder französischen Markt eine Marktzulassung (AMM) erhalten (L5121-8 Code de la santé publique).
Ein Arzneimittel ist (L 5111-1 des Code de la santé publique oder Richtlinie der Europäischen Gemeinschaft 65/65/EWG vom 26. Januar 1965, Artikel 1):
- eine Substanz oder Zusammensetzung, die als heilend oder vorbeugend gegen Krankheiten dargestellt wird, oder
- eine Substanz oder Zusammensetzung, die zur Erstellung einer medizinischen Diagnose verabreicht werden kann, oder eine Substanz oder Zusammensetzung, die zur Wiederherstellung, Korrektur oder Veränderung von Körperfunktionen verabreicht werden kann.
Hauptphasen der Entwicklung eines Arzneimittels
Allgemein betrachtet sind dies die Hauptphasen im Lebenszyklus eines Arzneimittels, von der Entwicklung bis zur Vermarktung.
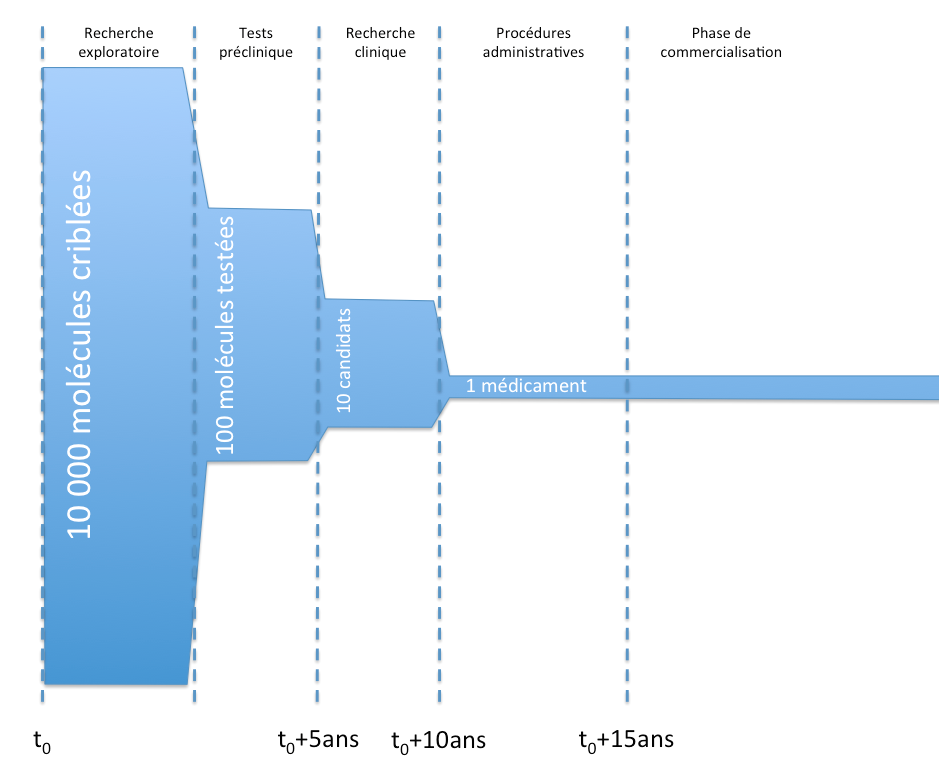 Lebenszyklus eines Arzneimittels und Marktzulassung
Lebenszyklus eines Arzneimittels und MarktzulassungDas Ziel der präklinischen Studien besteht insbesondere darin, die Verträglichkeit der Moleküle in vitro bei Tieren zu überprüfen, toxikologische Tests durchzuführen und die Pharmakokinetik der Moleküle zu untersuchen (grob gesagt, zu prüfen, wie schnell die Moleküle wirken, aufgenommen oder ausgeschieden werden).
Die klinische Forschung gliedert sich in drei Hauptphasen:
- Phase I:
- Versuche an menschlichen Patienten, an einer kleinen Kohorte gesunder Freiwilliger.
- Diese Versuche zielen insbesondere darauf ab, die Verträglichkeit der Moleküle bei den Patienten zu testen.
- Phase II:
- Versuche an menschlichen Patienten, an einer größeren Kohorte kranker Freiwilliger (ohne weitere Pathologien).
- Diese Versuche zielen insbesondere darauf ab, das Nutzen-Risiko-Verhältnis der Moleküle zu bestimmen und die wirksamen Dosen festzulegen.
- Phase III:
- Versuche an menschlichen Patienten, an einer großen Kohorte kranker Freiwilliger unter realen Bedingungen.
- Diese Versuche zielen insbesondere darauf ab, die Wirksamkeit (Studie im « Doppelblindverfahren« ) und die langfristige Sicherheit zu überprüfen.
Während der Vermarktungsphase wird ein Mechanismus der « Pharmakovigilanz » (L5121-22 des Code de la santé publique und folgende) eingerichtet: Mögliche Nebenwirkungen werden von Gesundheitsfachkräften erfasst und können zu Beobachtungsstudien führen.
Darüber hinaus können interventionelle Studien (sogenannte Phase IV) durchgeführt werden, insbesondere wenn die Marktzulassung dies aufgrund der geringen Anzahl von Patienten während der klinischen Studien verlangt (L5121-8-1 Code de la santé publique).
Schließlich können während der Vermarktungsphase sekundäre klinische Studien auf Initiative des Herstellers durchgeführt werden, um sein Arzneimittel bei einer anderen Erkrankung, einer neuen Bevölkerungsgruppe oder im Rahmen einer neuen therapeutischen Strategie zu testen, um eine neue Indikation zuzulassen.
Verfahren und Modalitäten
Verschiedene mögliche Verfahren
Um eine Marktzulassung zu erhalten, können verschiedene Wege gewählt werden:
- nationales Verfahren:
- gilt selbstverständlich nur für ein einziges Land
- zentralisiertes Verfahren: gilt für 27 europäische Länder; kann für bestimmte Arzneimittel (z. B. Biotechnologien oder neue Substanzen in bestimmten Bereichen wie Krebs oder HIV) verpflichtend sein
- Verfahren der gegenseitigen Anerkennung: Eine in einem Referenzland erhaltene Marktzulassung kann von anderen Ländern anerkannt werden
- dezentralisiertes Verfahren: ähnlich dem Verfahren der gegenseitigen Anerkennung, jedoch wurde noch keine Marktzulassung erteilt. Die Unterlagen werden in allen Ländern eingereicht, aber nur von einem Referenzland geprüft.
Dossier
Die Anmeldung eines Zulassungsdossiers erfolgt bei den zuständigen Behörden (französisch, europäisch usw.) in einem glücklicherweise harmonisierten Format (CTD-Format).
Das Zulassungsdossier enthält insbesondere folgende Angaben:
- administrative (abhängig von den zuständigen Behörden);
- zur Qualität (Herstellungsverfahren, verwendete Rohstoffe, Stabilität des Produkts usw.);
- zu präklinischen Studien;
- zur klinischen Forschung;
- zu den Angaben, die auf den Verpackungen und der Packungsbeilage für Patienten erscheinen müssen (im Anhang).
Entscheidungsfindung
Auf dieser Grundlage können die zuständigen Behörden die Zulassung auf der Basis von Qualitäts-, Wirksamkeits- und Sicherheitsinformationen des Arzneimittels erteilen: Eine Nutzen-Risiko-Bewertung wird dann unter Berücksichtigung anderer verfügbarer Behandlungen erstellt (L5121-9 Code de la santé publique).
Die Entscheidung ist wichtig, da sie die Bezeichnung, die Form des Arzneimittels, die Verabreichungsart, die Dosierung, die therapeutischen Indikationen, den Verschreiber und die Behandlungsdauer festlegt. Alle diese Elemente bestimmen indirekt den Preis und damit die Rentabilität des Arzneimittels.
Schutz der Zulassungsdaten
Grundsatz
Das Zulassungsdossier ist für einen bestimmten Zeitraum, den sogenannten „Daten-Schutzzeitraum“, nicht öffentlich zugänglich.
Während des „Daten-Schutzzeitraums“ dürfen die Zulassungsbehörden:
- weder das Dossier offenlegen
- noch einem anderen Unternehmen gestatten, sich auf eine Zulassung eines Dritten zu beziehen, um eine Genehmigung zu erhalten.
Die Dauer des „Daten-Schutzzeitraums“ beträgt 8 + 2 + 1 Jahre (Verordnung (EG) Nr. 726/2004, Art. 14(11)):
- 8 Jahre Schutz werden gewährt, in denen niemand Zugang zum Zulassungsdossier erhalten oder sich darauf beziehen darf (R5121-28 Code de la santé publique);
- 2 Jahre zusätzlicher Schutz, in denen es möglich ist, sich auf das Zulassungsdossier zu beziehen, jedoch keine Zulassung auf dieser Grundlage erteilt werden kann (L5121-10-1 Code de la santé publique);
- 1 Jahr zusätzlicher Schutz (zusätzlich zu den vorherigen 2 Jahren), wenn während der 8-Jahres-Periode eine neue therapeutische Indikation genehmigt wird, die einen bedeutenden Vorteil bietet (L5121-10-1 Code de la santé publique).
Ende des Schutzes des Zulassungsdossiers und Bestehen eines Patents
Wenn das Zulassungsdossier zugänglich ist und ein Dritter auf dieser Grundlage eine neue Zulassung erhält (Ende der 8+2+1-Jahres-Periode), bedeutet dies nicht, dass keine Verletzung vorliegt, falls ein Patent existiert und in Kraft ist.
Diese beiden Problemstellungen sind klar zu unterscheiden.
Schutz und Erweiterung des Sortiments
Wenn nach Erteilung einer Zulassung eine neue therapeutische Form oder eine neue Dosierung genehmigt wird (durch eine sekundäre klinische Studie), bedeutet dies nicht, dass eine neue Zulassung erteilt wird.
Eine Sortimentserweiterung ermöglicht somit keine Verlängerung der Schutzdauer einer Zulassung (R5121-41-1 Code de la santé publique oder Richtlinie 2001/83/EG des Europäischen Parlaments und des Rates vom 6. November 2001, Art. 6.1).
Schutz und seltene Erkrankung
Eine seltene Erkrankung betrifft nicht mehr als 5 Personen pro 10.000 in der Gemeinschaft (oder die Vermarktung eines Arzneimittels generiert keine ausreichenden Gewinne, um eine Investition zu rechtfertigen), und es gibt noch keine zugelassenen Arzneimittel (zumindest nicht mit vergleichbarer Wirksamkeit) (Verordnung (EG) Nr. 141/2000 des Europäischen Parlaments und des Rates vom 16. Dezember 1999, Artikel 3(1)).
Wird eine Zulassung für eine seltene Erkrankung beantragt, so besteht die Garantie, dass keine weitere Zulassung (auch keine Zulassungserweiterung) für ein ähnliches Arzneimittel erteilt wird (Verordnung (EG) Nr. 141/2000 des Europäischen Parlaments und des Rates vom 16. Dezember 1999, Artikel 8.1) für 10 Jahre.
Diese Frist von 10 Jahren kann auf 6 Jahre verkürzt werden, wenn vor Ablauf des 5. Jahres festgestellt wird, dass die Erkrankung keine seltene Erkrankung mehr im zuvor definierten Sinne ist (Verordnung (EG) Nr. 141/2000 des Europäischen Parlaments und des Rates vom 16. Dezember 1999, Artikel 8.2).
Dennoch kann diese 10-jährige Exklusivität reduziert werden, wenn (Verordnung (EG) Nr. 141/2000 des Europäischen Parlaments und des Rates vom 16. Dezember 1999, Artikel 8.2):
- der Inhaber zustimmt;
- der Inhaber sein Arzneimittel nicht in ausreichender Menge herstellen kann;
- ein neuer Antragsteller nachweist, dass sein Arzneimittel sicherer, wirksamer oder klinisch überlegen ist.
Schutz und seltene pädiatrische Erkrankung
Die in dem vorherigen Abschnitt genannte Frist von 10 Jahren für seltene Erkrankungen wird auf 12 Jahre verlängert, wenn der Zulassungsantrag Ergebnisse aller gemäß einem genehmigten pädiatrischen Prüfkonzept durchgeführten Studien umfasst (Verordnung (EG) Nr. 1901/2006 des Europäischen Parlaments und des Rates vom 12. Dezember 2006, Art. 37).
Es ist zu beachten, dass bei Vorliegen eines ergänzenden Schutzzertifikats (SPC) besondere Bestimmungen für pädiatrische Studien gelten (siehe unten).
Preisfestsetzung und Erstattung
Preisfestsetzung
Die Festsetzung des Arzneimittelpreises erfolgt im Rahmen von Verhandlungen zwischen den Herstellern und dem Wirtschaftsausschuss für Gesundheitsprodukte (CEPS); kommt keine Einigung zustande, setzt der CEPS den Preis einseitig fest. Der Preis wird durch ministerielle Verordnung festgelegt (Artikel L5123-1 des französischen Sozialgesetzbuchs) und berücksichtigt:
- die Preise anderer Arzneimittel mit gleicher therapeutischer Zielsetzung;
- die geplanten Verkaufsmengen;
- die Anwendungsbedingungen;
- den Fortschritt gegenüber bestehenden Behandlungen (oder ASMR, Verbesserung des medizinischen Nutzens).
Erstattung
In Abhängigkeit vom medizinischen Nutzen (SMR) wird entschieden, ob das Arzneimittel ganz oder teilweise von der gesetzlichen Krankenversicherung übernommen wird. Andere Arzneimittel werden bei dieser Bewertung nicht berücksichtigt.
Die endgültige Entscheidung über die Aufnahme in die Erstattungsliste obliegt den Ministern für Gesundheit und Sozialversicherung (Artikel L162-17-2-1 des französischen Sozialversicherungsgesetzbuchs).
Der SMR kann sein:
- erheblich: Erstattung von 65 % bis 100 % (der Höchstbetrag wird für unersetzliche und kostspielige Arzneimittel gewährt);
- mäßig: Erstattung zu 30 %;
- gering: Erstattung zu 15 %;
- unzureichend: nicht erstattet.
Generika und Biosimilars
Definition
Ein Generikum ist ein Arzneimittel mit der gleichen qualitativen und quantitativen Zusammensetzung an Wirkstoffen und der gleichen Darreichungsform wie ein Referenzarzneimittel (Artikel L5121-1 des französischen Sozialgesetzbuchs, 5° a oder Richtlinie 2001/83/EG des Europäischen Parlaments und des Rates vom 6. November 2001, Art. 10.2 b).
Darüber hinaus muss die Bioäquivalenz zwischen dem Generikum und dem Referenzarzneimittel nachgewiesen sein.
Vereinfachtes Verfahren für die Zulassung
Der Antrag auf Zulassung für ein Generikum ist vereinfacht.
Somit ist der Antragsteller nicht verpflichtet, folgende Ergebnisse vorzulegen:
- pharmakologische und toxikologische Prüfungen;
- klinische Studien.
Erforderlich sind lediglich die pharmazeutischen Daten, die der Qualität der Ausgangsstoffe und dem Herstellungsverfahren entsprechen.
Darüber hinaus muss nachgewiesen werden, dass das Generikum einem Arzneimittel ähnlich ist, das seit mindestens 10 Jahren eine Zulassung erhalten hat (Richtlinie 2001/83/EG des Europäischen Parlaments und des Rates vom 6. November 2001, Art. 10.1), und zwar durch eine Bioäquivalenzstudie (d. h. gleiches Verhalten im Organismus: Absorption, Verteilung, Metabolismus und Elimination).
Mögliche Unterschiede
Es ist daher möglich, eine unterschiedliche Darreichungsform (z. B. Sirup vs. Tablette) oder unterschiedliche Hilfsstoffe zu haben.
Ebenso ist es möglich, eine unterschiedliche therapeutische Indikation zu haben (d. h. Liste der Krankheiten, für die das Arzneimittel verwendet werden kann, Staatsrat, 23. Juli 2003, Nr. 246716).
Preisfestsetzung
Der Preis wird wie bei einem Standardarzneimittel festgelegt, jedoch wird dieser niedriger sein als der des Referenzarzneimittels, da die Forschungskosten geringer sind.
Ergänzende Schutzzertifikate oder SPC
Prinzip
Die SPC wurden geschaffen, um der besonders langen Dauer Rechnung zu tragen, die für die Erlangung der Zulassung erforderlich ist, und zu vermeiden, dass der Schutz verloren geht, bevor das Arzneimittel überhaupt auf den Markt gebracht wird.
Anwendbare Rechtsvorschriften?
Die Existenz des SPC ist vorgesehen durch:
- die Verordnung Nr. 1768/92 des Rates vom 18. Juni 1992:
- für die nach dem 2. Januar 1993 erteilten SPC (Artikel 23 der Verordnung), wenn das Patent ein Arzneimittel (Artikel 2 der Verordnung), dessen Herstellungsverfahren oder dessen Verwendung (Artikel 2 der Verordnung in Verbindung mit Artikel 1.c) betrifft;
- die Verordnung (EG) Nr. 1610/96 des Europäischen Parlaments und des Rates vom 23. Juli 1996:
- für die nach dem 8. Februar 1997 erteilten SPC (Artikel 23 der Verordnung), wenn das Patent ein Pflanzenschutzmittel betrifft (Artikel 2 der Verordnung);
- die Artikel L611-2 CPI und L611-3 CPI:
- für die vor dem 2. Januar 1993 erteilten SPC.
Soviel ist sicher, dass die Bestimmungen des CPI heute kaum noch anwendbar sind…
Eigenständiger Titel
Ein SPC ist ein eigenständiger Titel; es verlängert nicht die Laufzeit eines Patents (auch wenn die Existenz des Patents eine Voraussetzung ist): Es gewährt einen eigenen Schutz, der an dieses SPC gebunden ist.
Erteilungsvoraussetzungen für ein Zertifikat für ergänzende Schutzzertifikate (SPC)
Für die Erlangung eines SPC ist Folgendes erforderlich:
- dessen Beantragung
- Artikel 7 der Verordnung Nr. 1768/92 des Rates vom 18. Juni 1992 oder
- Artikel 7 der Verordnung (EG) Nr. 1610/96 des Europäischen Parlaments und des Rates vom 23. Juli 1996 ;
- das Vorliegen eines in Kraft befindlichen Patents (und nicht nur einer Anmeldung), das eines der zuvor genannten Elemente gemäß dem anwendbaren Text abdeckt;
- Artikel 3.a der Verordnung Nr. 1768/92 des Rates vom 18. Juni 1992 oder
- Artikel 3.a der Verordnung (EG) Nr. 1610/96 des Europäischen Parlaments und des Rates vom 23. Juli 1996 ;
- das Vorliegen einer gültigen Zulassung (AMM) für ein Arzneimittel-Spezialpräparat (d. h. den Handelsnamen eines Arzneimittels), das durch das Patent abgedeckt ist, oder falls das patentierte Arzneimittel oder Pflanzenschutzmittel Gegenstand einer Zulassung (AMM) war
- Artikel 3.b der Verordnung Nr. 1768/92 des Rates vom 18. Juni 1992 oder
- Artikel 3.b der Verordnung (EG) Nr. 1610/96 des Europäischen Parlaments und des Rates vom 23. Juli 1996) ;
- dass zuvor kein SPC beantragt wurde (
- Artikel 3.c der Verordnung Nr. 1768/92 des Rates vom 18. Juni 1992 oder
- Artikel 3.c der Verordnung (EG) Nr. 1610/96 des Europäischen Parlaments und des Rates vom 23. Juli 1996) ;
- dass diese Zulassung (AMM) die erste in der Gemeinschaft ist
- (Artikel 3.d der Verordnung Nr. 1768/92 des Rates vom 18. Juni 1992 oder
- Artikel 3.d der Verordnung (EG) Nr. 1610/96 des Europäischen Parlaments und des Rates vom 23. Juli 1996) ;
Schutzumfang
Der durch das SPC gewährte Schutz ist auf das durch die Zulassung (AMM) abgedeckte Spezialpräparat beschränkt (Artikel L611-3 CPI oder Artikel 4 der Verordnung Nr. 1768/92 des Rates vom 18. Juni 1992 oder Artikel 4 der Verordnung (EG) Nr. 1610/96 des Europäischen Parlaments und des Rates vom 23. Juli 1996).
Darüber hinaus ist es nicht möglich, ein SPC mit einem weiteren Schutzumfang (EuGH C-518/10 vom 25. November 2011) oder einem engeren Schutzumfang (EuGH C-322/10 vom 24. November 2011) als der Anspruch des betreffenden Patents zu erhalten (genaue Übereinstimmung): Wenn der Anspruch A1+A2 umfasst, ist es nicht möglich, ein SPC für A1+A2+A3 (selbst wenn die Zulassung für A1+A2+A3 erteilt wurde) oder nur für A1 zu erhalten.
Schutzdauer
Grundsatz
Das SPC ermöglicht einen Schutz mit einer Dauer, die nicht überschreiten darf (Artikel 13 der Verordnung Nr. 1768/92 des Rates vom 18. Juni 1992 oder Artikel 13 der Verordnung (EG) Nr. 1610/96 des Europäischen Parlaments und des Rates vom 23. Juli 1996):
- 5 Jahre ab dem Ablauf des Patents ;
- und die Zeitdifferenz zwischen dem Datum der ersten Zulassung (AMM) und dem Anmeldedatum des Patents, von der 5 Jahre abgezogen werden.
Zusammengefasst beträgt die durch das SPC gewährte Schutzdauer:
- wenn die Zulassung (AMM) mehr als 10 Jahre nach der Patentanmeldung erteilt wird: 5 Jahre ;
- wenn die Zulassung (AMM) schneller als in 10 Jahren erteilt wird, verkürzt sich die Dauer von 5 Jahren entsprechend.
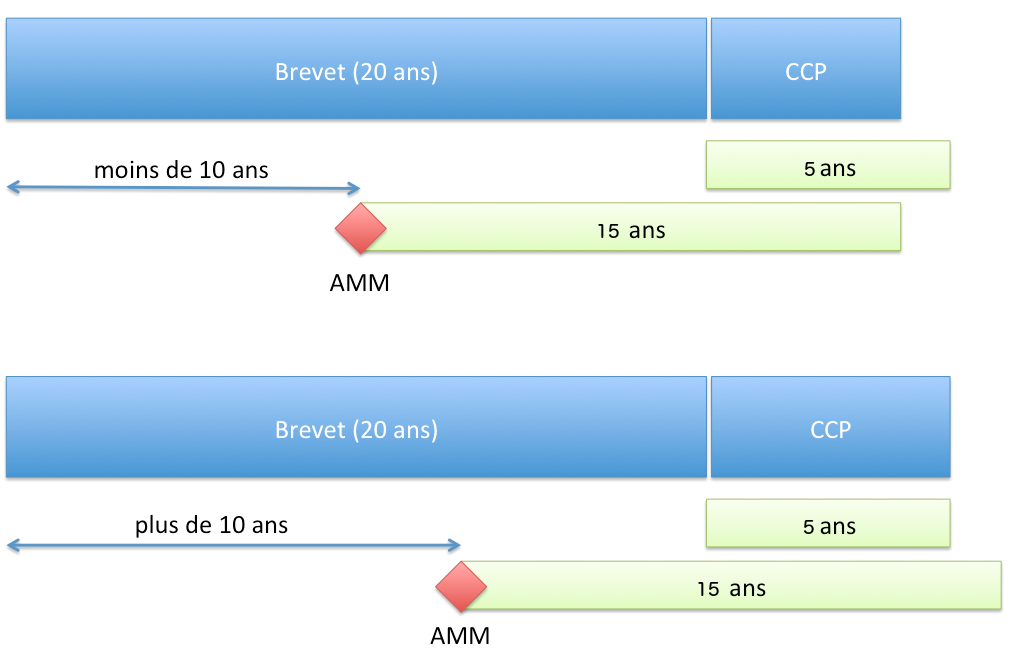 Schutzdauer durch ein SPC (EU-Verordnung)
Schutzdauer durch ein SPC (EU-Verordnung)Beginn der Frist
Die Frage, die sich stellte, war, worauf sich das « Datum der ersten Genehmigung für das Inverkehrbringen » bezog:
- das Datum der Entscheidung, die die Zulassung erteilt, oder
- das Datum der Bekanntgabe dieser Entscheidung oder
- das Datum der Erteilung.
Dies kann von Bedeutung sein…
Der EuGH hatte Gelegenheit, zu diesem Thema Stellung zu nehmen (EuGH C‑471/14, 6. Oktober 2015) und hat angegeben, dass das « Datum der ersten Genehmigung für das Inverkehrbringen » das Datum der Bekanntgabe dieser Entscheidung war.
Negative Laufzeit?
Aufgrund der zuvor genannten Berechnungsmethode ist es durchaus möglich, dass die Laufzeit eines Zertifikats für ergänzende Schutzzertifikate (SPC) negativ ist.
Auch wenn der Inhaber des Patents normalerweise nicht daran interessiert ist, ein SPC mit negativer Laufzeit zu erhalten, kann er dies ausnahmsweise wünschen, insbesondere wenn er eine unten erwähnte pädiatrische Verlängerung in Anspruch nehmen möchte.
Der EuGH hat angegeben, dass dies durchaus möglich ist (EuGH C‑125/10, 8. Dezember 2011).
Pädiatrische Verlängerung
Wenn der Inhaber eines SPC die Ergebnisse der Studien vorlegt, die gemäß einem pädiatrischen Prüfkonzept (teilweise oder vollständig für die Altersgruppe von 0 bis 18 Jahren) durchgeführt wurden, das in seiner Zulassungs- oder ergänzenden Zulassungsanmeldung genehmigt wurde, hat er Anspruch auf eine Verlängerung seines SPC um 6 Monate (Verordnung (EG) Nr. 1901/2006 des Europäischen Parlaments und des Rates vom 12. Dezember 2006, Art. 36.1).
Diese Verlängerung ist jedoch nicht möglich:
- wenn das Arzneimittel in der Zulassung als für eine pädiatrische seltene Krankheit bestimmt ausgewiesen ist (ein zusätzlicher Schutz der Zulassung ist bereits gewährt, siehe oben) (Verordnung (EG) Nr. 1901/2006 des Europäischen Parlaments und des Rates vom 12. Dezember 2006, Art. 36.4);
- wenn die Zulassung einen zusätzlichen einjährigen Schutz aufgrund einer neuen therapeutischen Indikation genießt (siehe oben) (Verordnung (EG) Nr. 1901/2006 des Europäischen Parlaments und des Rates vom 12. Dezember 2006, Art. 36.5).
Dans quel délai peut-on demander le CCP ? merci d’avance