
El procedimiento AMM: « Autorización de Comercialización »
¿Quién está afectado por la AMM?
Los medicamentos, antes de ser introducidos en el mercado europeo o español, deben recibir una AMM (L5121-8 código de la salud pública).
Un medicamento es (L 5111-1 del Código de la salud pública o directiva de la Comunidad Europea 65/65/CEE del 26 de enero de 1965, artículo 1):
- una sustancia o composición presentada como poseedora de propiedades curativas o preventivas respecto a enfermedades, o
- una sustancia o composición que pueda ser administrada con el fin de establecer un diagnóstico médico, o una sustancia o composición que pueda ser administrada con el fin de restaurar, corregir o modificar funciones orgánicas.
Principales etapas en el desarrollo de un medicamento
Desde un punto de vista general, estas son las principales etapas en la vida de un medicamento, desde su desarrollo hasta su comercialización.
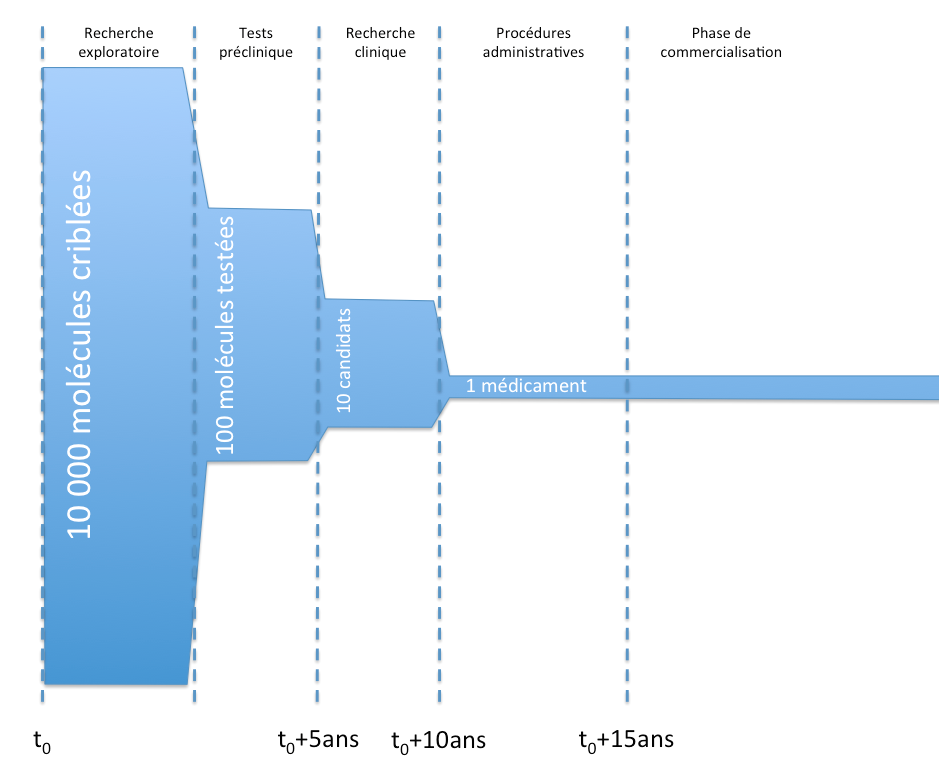 Vida de un medicamento y AMM
Vida de un medicamento y AMMEl objetivo de los ensayos preclínicos es, en particular, verificar la tolerancia de las moléculas in vitro en animales, realizar pruebas toxicológicas y estudiar la farmacocinética de las moléculas (en resumen, observar a qué velocidad actúan, son absorbidas o eliminadas las moléculas).
La investigación clínica se divide en tres grandes fases:
- fase I:
- ensayos en pacientes humanos, en una pequeña cohorte de voluntarios sanos.
- Estos ensayos tienen como objetivo, en particular, probar la tolerancia de las moléculas en los pacientes.
- fase II:
- ensayos en pacientes humanos, en una cohorte de mayor tamaño de voluntarios enfermos (pero sin otras patologías).
- Estos ensayos tienen como objetivo, en particular, establecer una relación beneficio/riesgo de la molécula y definir las dosis de eficacia.
- fase III:
- ensayos en pacientes humanos, en una cohorte de gran tamaño de voluntarios enfermos en condiciones reales.
- Estos ensayos tienen como objetivo, en particular, verificar la eficacia (estudio en « doble ciego« ) y la seguridad a largo plazo.
Durante la fase de comercialización, se implementa un mecanismo de « farmacovigilancia » (L5121-22 del código de la salud pública y siguientes): se recogen posibles efectos secundarios de los profesionales de la salud y pueden dar lugar a estudios observacionales.
Además, se pueden realizar estudios intervencionistas (denominados de fase IV) especialmente si la AMM lo exige debido al reducido número de pacientes durante los ensayos clínicos (L5121-8-1 código de la salud pública).
Por último, durante la fase de comercialización, se pueden llevar a cabo estudios clínicos secundarios por iniciativa del industrial para probar su medicamento en otra patología, una nueva población, en el marco de una nueva estrategia terapéutica, etc., con el fin de autorizar una nueva indicación.
Procedimientos y modalidades
Diferentes procedimientos posibles
Para obtener una AMM, es posible elegir diferentes vías:
- procedimiento nacional:
- solo es válido, por supuesto, para un único país.
- Procedimiento centralizado: cubre 27 países europeos; puede ser obligatorio para ciertos medicamentos (ej. biotecnologías o nuevas sustancias en ciertos ámbitos como cáncer o VIH); procedimiento de reconocimiento mutuo: una AMM obtenida en un país de referencia puede ser reconocida por los demás países; procedimiento descentralizado: similar al procedimiento de reconocimiento mutuo, pero sin que se haya obtenido aún ninguna AMM. Los expedientes se presentan en todos los países, pero solo son evaluados por un país de referencia.
Expediente
La solicitud de un expediente de AMM se realiza ante las autoridades competentes (francesa, europeas, etc.) en un formato que, afortunadamente, está armonizado (formato CTD).
El expediente de AMM incluye, en particular, las siguientes indicaciones:
- administrativas (depende de las autoridades competentes);
- sobre la calidad (proceso de fabricación, materia prima utilizada, estabilidad del producto, etc.);
- sobre los ensayos preclínicos;
- sobre la investigación clínica;
- sobre las menciones que deben figurar en los envases y el prospecto para uso de los pacientes (en anexo).
Toma de decisión
Sobre esta base, las autoridades competentes pueden conceder la AMM en función de la información sobre la calidad, la eficacia del medicamento y la seguridad: se establece entonces un balance beneficio/riesgo, teniendo en cuenta los demás tratamientos disponibles (L5121-9 Código de la salud pública).
La decisión es importante, ya que fija la denominación, la forma del medicamento, el modo de administración, la dosificación, las indicaciones terapéuticas, el prescriptor, la duración del tratamiento… El conjunto de estos elementos determinará indirectamente el precio y, por tanto, la rentabilidad del medicamento.
Protección de los datos del AMM
Principio
El expediente de AMM no es público durante un cierto período, denominado período de «protección de datos».
Durante el período de «protección de datos», las autoridades reguladoras no pueden:
- ni divulgar el expediente;
- ni autorizar a otra empresa a hacer referencia a una AMM de un tercero para obtener una autorización.
La duración del período de «protección de datos» es de 8 + 2 + 1 años (Reglamento CE n°726/2004, art. 14(11)):
- 8 años de protección, período durante el cual nadie puede acceder ni hacer referencia al expediente de AMM (R5121-28 Código de la salud pública);
- 2 años de protección adicional, período durante el cual es posible hacer referencia al expediente de AMM, pero durante el cual no es posible obtener una AMM sobre esta base (L5121-10-1 Código de la salud pública);
- 1 año de protección adicional (respecto a los 2 años anteriores), si durante el período de los 8 años se concede una nueva indicación terapéutica y esta aporta una ventaja importante (L5121-10-1 Código de la salud pública).
Fin de la protección del expediente de AMM y existencia de una patente
Si el expediente de AMM es accesible y un tercero obtiene una nueva AMM sobre esta base (fin del período 8+2+1), esto no significa que no sea infractor si existe una patente en vigor.
Es importante distinguir claramente estos dos aspectos.
Protección y extensión de gama
Si tras la obtención de una AMM se autoriza una nueva forma terapéutica o una nueva dosificación (mediante un estudio clínico secundario), esto no significa que se conceda una nueva AMM.
Así, una extensión de gama no permite ampliar la duración de la protección de una AMM (R5121-41-1 Código de la salud pública o Directiva 2001/83/CE del Parlamento Europeo y del Consejo de 6 de noviembre de 2001, art. 6.1).
Protección y enfermedad rara
Una enfermedad rara no afecta a más de 5 personas por cada 10 000 en la Comunidad (o que la comercialización de un medicamento no genere beneficios suficientes para justificar una inversión) y que aún no existan medicamentos autorizados (al menos, con una eficacia comparable) (Reglamento n°141/2000 del Parlamento Europeo y del Consejo, de 16 de diciembre de 1999, artículo 3(1)).
Si se solicita una AMM para una enfermedad rara, entonces existe una garantía de que no se concederá ninguna otra AMM (incluso una extensión de AMM) para un medicamento similar (Reglamento n°141/2000 del Parlamento Europeo y del Consejo, de 16 de diciembre de 1999, artículo 8.1) durante 10 años.
Este plazo de 10 años puede reducirse a 6 años, si se establece antes de finalizar el quinto año que la enfermedad ya no es una enfermedad rara según la definición anterior (Reglamento n°141/2000 del Parlamento Europeo y del Consejo, de 16 de diciembre de 1999, artículo 8.2).
No obstante, esta exclusividad de 10 años puede reducirse si (Reglamento n°141/2000 del Parlamento Europeo y del Consejo, de 16 de diciembre de 1999, artículo 8.2) :
- el titular da su acuerdo ;
- el titular no puede producir su medicamento en cantidad suficiente ;
- un nuevo solicitante demuestra que su medicamento es más seguro, más eficaz o clínicamente superior.
Protección y enfermedad rara pediátrica
El plazo de 10 años mencionado para las enfermedades raras en la sección anterior se amplía a 12 años si la solicitud de AMM incluye los resultados de todos los estudios realizados según un plan de investigación pediátrica aprobado (Reglamento CE n°1901/2006 del Parlamento Europeo y del Consejo, de 12 de diciembre de 2006, art 37).
Cabe señalar que, si existe un CCP, existen disposiciones particulares relativas a los estudios pediátricos (véase más abajo).
Fijación de precios y reembolso
Fijación de precios
La fijación del precio del medicamento se realiza mediante una negociación entre los industriales y el Comité Económico de Productos Sanitarios (o CEPS; en caso de desacuerdo, el CEPS fija unilateralmente el precio). El precio, establecido por orden ministerial (L5123-1 código de la salud pública), tiene en cuenta :
- los precios de otros medicamentos con la misma finalidad terapéutica ;
- los volúmenes de ventas previstos ;
- las condiciones de uso ;
- el progreso respecto a los tratamientos existentes (o ASMR, mejora del servicio médico prestado).
Reembolso
En función del servicio médico prestado (SMR), se decide si el medicamento es reembolsado, total o parcialmente, por la seguridad social. Los demás medicamentos no se tienen en cuenta en esta evaluación.
La decisión final de inclusión en el reembolso corresponde a los ministros encargados de Sanidad y de la Seguridad Social (L162-17-2-1 código de la seguridad social).
El SMR puede ser :
- importante : reembolso del 65 % al 100 % (el tipo máximo se concede a los medicamentos insustituibles y costosos) ;
- moderado : reembolso del 30 % ;
- bajo : reembolso del 15 % ;
- insuficiente : no reembolsado.
Genéricos y biosimilares
Definición
Un genérico es un medicamento que tiene la misma composición cualitativa y cuantitativa en principio activo y la misma forma farmacéutica que un medicamento de referencia (L5121-1 código de la salud pública, 5° a o Directiva 2001/83/CE del Parlamento Europeo y del Consejo, de 6 de noviembre de 2001, art 10.2 b).
Además, debe haberse demostrado la bioequivalencia entre el genérico y el medicamento de referencia.
Procedimiento simplificado para la AMM
La solicitud de AMM para un genérico está simplificada.
Así, el solicitante no está obligado a aportar resultados:
- de ensayos farmacológicos y toxicológicos;
- de ensayos clínicos.
Solo se requieren los datos farmacéuticos correspondientes a la calidad de las materias primas y al proceso de fabricación.
Por otra parte, es necesario demostrar que el genérico es similar a un medicamento que haya obtenido una AMM desde al menos 10 años (Directiva 2001/83/CE del Parlamento Europeo y del Consejo de 6 de noviembre de 2001, art 10.1) mediante un estudio de bioequivalencia (es decir, comportamiento equivalente en el organismo: absorción, distribución, metabolismo y eliminación).
Diferencias posibles
Por tanto, es posible tener una presentación diferente (ej. jarabe vs. píldora) o excipientes diferentes.
También es posible tener una indicación terapéutica diferente (es decir, lista de patologías para las que el medicamento puede ser utilizado, Consejo de Estado, 23 de julio de 2003, n°246716).
Determinación del precio
El precio se determina como para un medicamento estándar, pero este será más bajo que el del medicamento de referencia, ya que los costes de investigación son menores.
Los Certificados Complementarios de Protección o CCP
Principio
Los CCP se crearon para tener en cuenta la duración particularmente larga necesaria para la obtención de la AMM y evitar que la protección se pierda incluso antes del inicio de la comercialización del medicamento.
¿Los textos aplicables?
La existencia del CCP está prevista por:
- el Reglamento n°1768/92 del Consejo de 18 de junio de 1992:
- para los CCP concedidos después del 2 de enero de 1993 (artículo 23 del reglamento) si la patente se refiere a un medicamento (artículo 2 del reglamento), su procedimiento de obtención o su uso (artículo 2 del reglamento junto con el artículo 1.c);
- el Reglamento (CE) n° 1610/96 del Parlamento Europeo y del Consejo de 23 de julio de 1996:
- para los CCP concedidos después del 8 de febrero de 1997 (artículo 23 del reglamento) si la patente se refiere a un producto fitosanitario (artículo 2 del reglamento);
- los artículos L611-2 CPI y L611-3 CPI:
- para los CCP concedidos antes del 2 de enero de 1993.
Dicho de otro modo, las disposiciones del CPI ya no son muy utilizables hoy en día…
Título autónomo
Un CCP es un título autónomo, no prolonga la duración de una patente (aunque la existencia de la patente sea un prerrequisito): confiere una protección propia y vinculada a dicho CCP.
Condición de concesión de los CCP
Para obtener un CCP, es necesario:
- solicitarlo
- disponer de una patente en vigor (y no una solicitud) que cubra uno de los elementos mencionados anteriormente en función del texto aplicable;
- artículo 3.a del Reglamento n°1768/92 del Consejo de 18 de junio de 1992 o
- artículo 3.a del Reglamento (CE) n° 1610/96 del Parlamento Europeo y del Consejo de 23 de julio de 1996;
- disponer de una AMM en vigor que cubra una especialidad de ese medicamento (es decir, el nombre de marca comercial de un medicamento) cubierto por la patente o si el medicamento o el producto fitosanitario patentado ha sido objeto de una AMM
- artículo 3.b del Reglamento n°1768/92 del Consejo de 18 de junio de 1992 o
- artículo 3.b del Reglamento (CE) n° 1610/96 del Parlamento Europeo y del Consejo de 23 de julio de 1996);
- no haber solicitado ya un CCP (
- artículo 3.c del Reglamento n°1768/92 del Consejo de 18 de junio de 1992 o
- artículo 3.c del Reglamento (CE) n° 1610/96 del Parlamento Europeo y del Consejo de 23 de julio de 1996);
- que dicha AMM sea la primera en la Comunidad
-
(
- artículo 3.d del Reglamento n°1768/92 del Consejo de 18 de junio de 1992 o
- artículo 3.d del Reglamento (CE) n° 1610/96 del Parlamento Europeo y del Consejo de 23 de julio de 1996);
Alcance de la protección
La protección conferida por el CCP se limita a la especialidad cubierta por la AMM (L611-3 CPI o artículo 4 del Reglamento n°1768/92 del Consejo de 18 de junio de 1992 o el artículo 4 del Reglamento (CE) n° 1610/96 del Parlamento Europeo y del Consejo de 23 de julio de 1996).
Por otra parte, no es posible obtener un CCP más amplio (CJUE C-518/10 de 25 de noviembre de 2011) o más restringido (CJUE C-322/10 de 24 de noviembre de 2011) que la reivindicación de la patente invocada (concordancia exacta): si la reivindicación se refiere a A1+A2, no es posible obtener un CCP sobre A1+A2+A3 (aun cuando la AMM se hubiera concedido para A2+A2+A3) o sobre A1 solo.
Duración de la protección
Principio
El CCP permite obtener una protección de una duración que no puede exceder (artículo 13 del Reglamento n°1768/92 del Consejo de 18 de junio de 1992 o el artículo 13 del Reglamento (CE) n° 1610/96 del Parlamento Europeo y del Consejo de 23 de julio de 1996):
- 5 años a partir de la expiración de la patente;
- y la diferencia de tiempo entre la fecha de la primera AMM y la fecha de solicitud de la patente a la que se resta 5 años.
En resumen, la duración de la protección otorgada por el CCP es de:
- si la AMM se concede más de 10 años después de la solicitud de la patente: 5 años;
- si la AMM se concede en un plazo inferior a 10 años, la duración de 5 años se reduce en la misma medida.
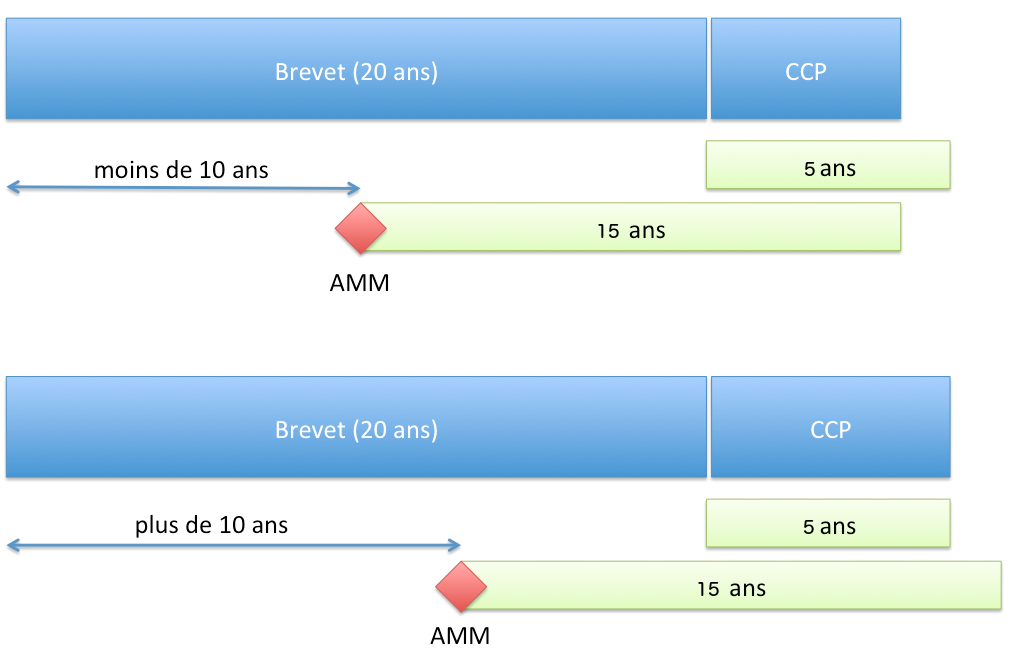 Duración de la protección conferida por un CCP (Reglamento UE)
Duración de la protección conferida por un CCP (Reglamento UE)Inicio del plazo
La cuestión que se planteó es a qué hacía referencia la « fecha de la primera autorización de comercialización« :
- la fecha de la decisión por la que se concede la AC
- la fecha de la notificación de dicha decisión o
- la fecha de concesión.
Esto puede tener su importancia…
El TJUE ha tenido ocasión de pronunciarse sobre el tema (CJUE C‑471/14, 6 octubre 2015) e indicó que la « fecha de la primera autorización de comercialización » era la fecha de la notificación de dicha decisión.
¿Duración negativa?
Debido al método de cálculo mencionado anteriormente, es perfectamente posible que la duración de un CCP sea negativa.
Aunque habitualmente el titular de la patente no esté interesado en obtener un CCP de duración negativa, puede excepcionalmente desearlo, en particular si pretende beneficiarse de una prórroga pediátrica mencionada a continuación.
El TJUE indicó que esto era perfectamente posible (CJUE C‑125/10, 8 diciembre 2011).
Prórroga pediátrica
Si el titular de un CCP aporta los resultados de los estudios realizados conforme a un plan de investigación pediátrica (en parte o en su totalidad en el tramo de 0 a 18 años) aprobado en su solicitud de AC o de AC complementaria, tendrá derecho a una prórroga de 6 meses de su CCP (Reglamento CE n°1901/2006 del Parlamento Europeo y del Consejo de 12 de diciembre de 2006, art 36.1).
No obstante, esta prórroga no es posible:
- si el medicamento está designado en la AC como dirigido a una enfermedad rara pediátrica (ya se concede una protección adicional de la AC, véase supra) (Reglamento CE n°1901/2006 del Parlamento Europeo y del Consejo de 12 de diciembre de 2006, art 36.4);
- si la AC se beneficia de una protección adicional de un año concedida en caso de nueva indicación terapéutica (véase supra) (Reglamento CE n°1901/2006 del Parlamento Europeo y del Consejo de 12 de diciembre de 2006, art 36.5).
Dans quel délai peut-on demander le CCP ? merci d’avance