
La procedura AIC: « Autorizzazione all’Immissione in Commercio »
Chi è interessato dall’AIC?
I medicinali, prima di essere introdotti sul mercato europeo o italiano, devono ottenere un’AIC (L5121-8 codice della sanità pubblica).
Un medicinale è (L 5111-1 del Codice della sanità pubblica o direttiva della Comunità europea 65/65/CEE del 26 gennaio 1965, articolo 1):
- una sostanza o composizione presentata come avente proprietà curative o preventive nei confronti di malattie, o
- una sostanza o composizione che può essere somministrata per stabilire una diagnosi medica, o una sostanza o composizione che può essere somministrata per ripristinare, correggere o modificare funzioni organiche.
Principali fasi della concezione di un medicinale
Da un punto di vista generale, ecco le principali fasi della vita di un medicinale, dalla concezione alla commercializzazione.
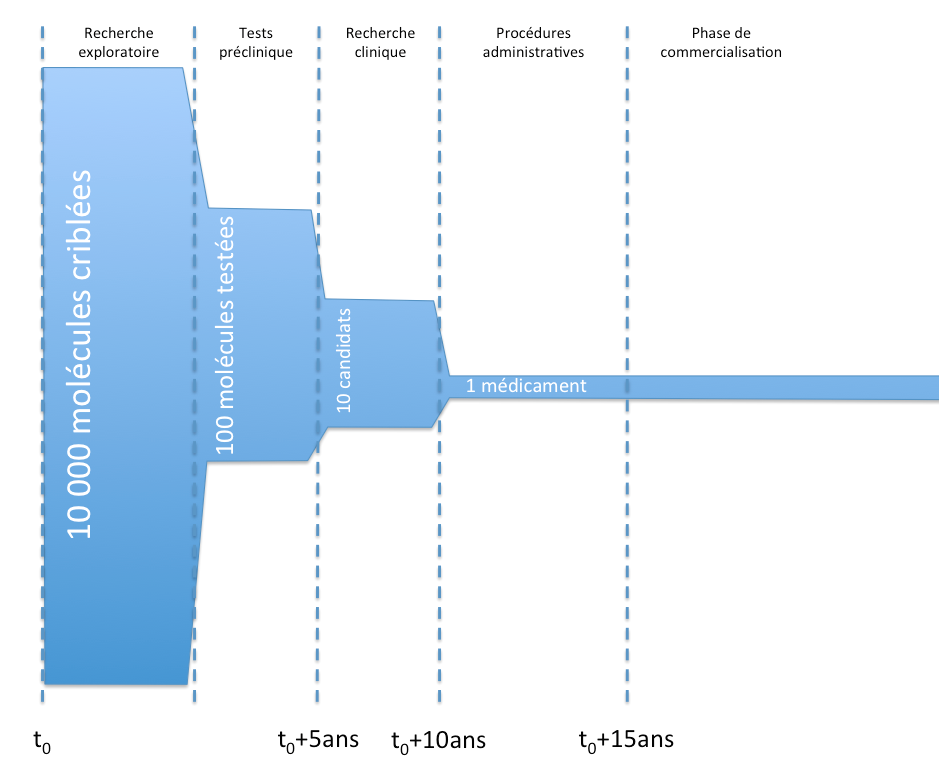 Vita di un medicinale e AIC
Vita di un medicinale e AICL’obiettivo degli studi preclinici è, in particolare, verificare la tolleranza delle molecole in vitro sugli animali, effettuare test tossicologici e studiare la farmacocinetica delle molecole (in sintesi, osservare la velocità con cui le molecole agiscono, vengono assorbite o eliminate).
La ricerca clinica si divide in tre grandi fasi:
- fase I:
- studi su pazienti umani, su una piccola coorte di volontari sani.
- Questi studi mirano, in particolare, a testare la tolleranza delle molecole nei pazienti.
- fase II:
- studi su pazienti umani, su una coorte più ampia di volontari malati (ma senza altre patologie).
- Questi studi mirano, in particolare, a stabilire un rapporto beneficio/rischio della molecola e a definire le dosi di efficacia.
- fase III:
- studi su pazienti umani, su una coorte ampia di volontari malati in condizioni reali.
- Questi studi mirano, in particolare, a verificare l’efficacia (studio in « doppio cieco« ) e la sicurezza a lungo termine.
Durante la fase di commercializzazione, viene attuato un meccanismo di « farmacovigilanza » (L5121-22 del codice della sanità pubblica e seguenti): vengono raccolti eventuali effetti collaterali dai professionisti della salute e possono portare a studi osservazionali.
Inoltre, possono essere condotti studi interventistici (detti di fase IV), in particolare se l’AIC lo richiede a causa del ridotto numero di pazienti durante gli studi clinici (L5121-8-1 codice della sanità pubblica).
Infine, durante la fase di commercializzazione, possono essere condotti studi clinici secondari su iniziativa dell’industriale per testare il proprio medicinale su un’altra patologia, una nuova popolazione, nell’ambito di una nuova strategia terapeutica, ecc. al fine di ottenere l’autorizzazione per una nuova indicazione.
Procedure e modalità
Diverse procedure possibili
Per ottenere un’AIC, è possibile scegliere diverse vie:
- procedura nazionale:
- vale ovviamente solo per un singolo paese.
- Procedura centralizzata: copre 27 paesi europei; può essere obbligatoria per alcuni medicinali (es. biotecnologie o nuove sostanze in alcuni ambiti come cancro o HIV); procedura di mutuo riconoscimento: un’AIC ottenuta in un paese di riferimento può essere riconosciuta dagli altri paesi; procedura decentrata: simile alla procedura di mutuo riconoscimento, ma nessuna AIC è ancora stata ottenuta. I dossier vengono depositati in tutti i paesi, ma esaminati solo da un paese di riferimento.
Fascicolo
Il deposito di un fascicolo AIC viene effettuato presso le autorità competenti (francesi, europee, ecc.) in un formato che fortunatamente è armonizzato (formato CTD).
Il fascicolo AIC comprende in particolare le seguenti indicazioni:
- amministrative (ciò dipende dalle autorità competenti);
- sulla qualità (processo di fabbricazione, materia prima utilizzata, stabilità del prodotto, ecc.);
- sugli studi preclinici;
- sulla ricerca clinica;
- sulle informazioni che devono figurare sugli imballaggi e sul foglietto illustrativo per l’uso da parte dei pazienti (in allegato).
Presa di decisione
Su questa base, le autorità competenti possono concedere l’AIC sulla base delle informazioni relative alla qualità, all’efficacia del medicinale e alla sicurezza: viene quindi stabilito un rapporto beneficio/rischio tenendo conto degli altri trattamenti disponibili (L5121-9 Codice della sanità pubblica).
La decisione è importante, poiché stabilisce la denominazione, la forma del medicinale, la modalità di somministrazione, il dosaggio, le indicazioni terapeutiche, il prescrittore, la durata del trattamento… L’insieme di questi elementi determina indirettamente il prezzo e quindi la redditività del medicinale.
Protezione dei dati dell’AIC
Principio
Il fascicolo dell’AIC non è pubblico per un determinato periodo, detto periodo di « protezione dei dati« .
Durante il periodo di « protezione dei dati« , le autorità regolatorie non possono:
- né divulgare il fascicolo
- né autorizzare un’altra società a fare riferimento a un’AIC di terzi per ottenere un’autorizzazione.
La durata del periodo di « protezione dei dati » è di 8 + 2 + 1 anni (Regolamento CE n. 726/2004, art. 14(11)):
- 8 anni di protezione sono concessi, periodo durante il quale nessuno può accedere o fare riferimento al fascicolo AIC (R5121-28 Codice della sanità pubblica);
- 2 anni di protezione supplementare, periodo durante il quale è possibile fare riferimento al fascicolo AIC, ma durante il quale non è possibile ottenere un’AIC su questa base (L5121-10-1 Codice della sanità pubblica);
- 1 anno di protezione supplementare (rispetto ai 2 anni precedenti), se durante il periodo degli 8 anni viene concessa una nuova indicazione terapeutica e questa apporta un vantaggio importante (L5121-10-1 Codice della sanità pubblica).
Fine della protezione del fascicolo AIC ed esistenza di un brevetto
Se il fascicolo AIC è accessibile e un terzo ottiene una nuova AIC su questa base (fine del periodo 8+2+1), ciò non significa che non sarà contraffattore se esiste un brevetto valido e in vigore.
È necessario distinguere chiaramente queste due problematiche.
Protezione ed estensione della gamma
Se dopo l’ottenimento di un’AIC viene autorizzata una nuova forma terapeutica o un nuovo dosaggio (tramite uno studio clinico secondario), ciò non significa che venga concessa una nuova AIC.
Pertanto, un’estensione della gamma non consente di prolungare la durata di protezione di un’AIC (R5121-41-1 Codice della sanità pubblica o Direttiva 2001/83/CE del Parlamento Europeo e del Consiglio del 6 novembre 2001, art 6.1).
Protezione e malattia rara
Una malattia rara non colpisce più di 5 persone su 10.000 nella Comunità (o che la commercializzazione di un medicinale non generi profitti sufficienti a giustificare un investimento) e che non esistano ancora medicinali autorizzati (almeno, con un’efficacia comparabile) (Regolamento n°141/2000 del Parlamento europeo e del Consiglio, del 16 dicembre 1999, articolo 3(1)).
Se viene richiesta un’AIC per una malattia rara, allora esiste una garanzia che nessun’altra AIC (nemmeno un’estensione di AIC) sarà concessa per un medicinale simile (Regolamento n°141/2000 del Parlamento europeo e del Consiglio, del 16 dicembre 1999, articolo 8.1) per 10 anni.
Tale periodo di 10 anni può essere ridotto a 6 anni, se viene stabilito prima della fine del 5° anno che la malattia non è più una malattia rara come definita in precedenza (Regolamento n°141/2000 del Parlamento europeo e del Consiglio, del 16 dicembre 1999, articolo 8.2).
Tuttavia, questa esclusività di 10 anni può essere ridotta se (Regolamento n°141/2000 del Parlamento europeo e del Consiglio, del 16 dicembre 1999, articolo 8.2) :
- il titolare dà il proprio consenso ;
- il titolare non è in grado di produrre il proprio medicinale in quantità sufficiente ;
- un nuovo richiedente dimostra che il proprio medicinale è più sicuro, più efficace o clinicamente superiore.
Protezione e malattia rara pediatrica
Il periodo di 10 anni menzionato per le malattie rare nella sezione precedente è portato a 12 anni se la domanda di AIC comprende i risultati di tutti gli studi realizzati secondo un piano di indagine pediatrica approvato (Regolamento CE n°1901/2006 del Parlamento Europeo e del Consiglio del 12 dicembre 2006, art 37).
Va notato che, se esiste un CCP, esistono disposizioni particolari riguardanti gli studi pediatrici (vedi sotto).
Determinazione dei prezzi e rimborso
Determinazione dei prezzi
La determinazione del prezzo del medicinale viene effettuata durante una negoziazione tra gli industriali e il Comitato Economico dei Prodotti della Salute (o CEPS, in mancanza di accordo, il CEPS fissa unilateralmente il prezzo). Il prezzo, fissato con decreto ministeriale (L5123-1 codice della sanità pubblica), tiene conto :
- dei prezzi degli altri medicinali con la stessa indicazione terapeutica ;
- dei volumi di vendita previsti ;
- delle condizioni di utilizzo ;
- del progresso rispetto ai trattamenti esistenti (o ASMR, miglioramento del servizio medico reso).
Rimborso
In funzione del servizio medico reso (SMR), viene deciso se il medicinale è, in tutto o in parte, rimborsato dal servizio sanitario nazionale. Gli altri medicinali non sono presi in considerazione in questa valutazione.
La decisione finale di iscrizione al rimborso spetta ai ministri competenti per la Salute e la Sicurezza sociale (L162-17-2-1 codice della sicurezza sociale).
Il SMR può essere :
- importante : rimborso dal 65 % al 100 % (la percentuale massima è concessa ai medicinali insostituibili e costosi) ;
- moderato : rimborso al 30 % ;
- scarso : rimborso al 15 % ;
- insufficiente : non rimborsato.
Generici e bio-similare
Definizione
Un generico è un medicinale che ha la stessa composizione qualitativa e quantitativa in principi attivi e la stessa forma farmaceutica di un medicinale di riferimento (L5121-1 codice della sanità pubblica, 5° a o Direttiva 2001/83/CE del Parlamento Europeo e del Consiglio del 6 novembre 2001, art 10.2 b).
È inoltre necessario che la bioequivalenza tra il generico e il medicinale di riferimento sia stata dimostrata.
Procedura semplificata per l’AIC
La domanda di AIC per un generico è semplificata.
Pertanto, il richiedente non è tenuto a fornire i risultati:
- delle prove farmacologiche e tossicologiche;
- delle prove cliniche.
Sono richiesti unicamente i dati farmaceutici relativi alla qualità delle materie prime e al processo di fabbricazione.
Inoltre, è necessario dimostrare che il generico è simile a un medicinale che ha ottenuto un’AIC da almeno 10 anni (Direttiva 2001/83/CE del Parlamento Europeo e del Consiglio del 6 novembre 2001, art 10.1) tramite uno studio di bioequivalenza (ossia comportamento equivalente nell’organismo: assorbimento, distribuzione, metabolismo ed eliminazione).
Differenze possibili
È quindi possibile avere una presentazione diversa (es. sciroppo vs. pillola) o eccipienti diversi.
È inoltre possibile avere un’indicazione terapeutica diversa (ossia l’elenco delle patologie per le quali il medicinale può essere utilizzato, Consiglio di Stato, 23 luglio 2003, n°246716).
Determinazione del prezzo
Il prezzo è determinato come per un medicinale standard, ma sarà inferiore rispetto al medicinale di riferimento, poiché i costi di ricerca sono più bassi.
I Certificati Complementari di Protezione o CCP
Principio
I CCP sono stati creati per tener conto della durata particolarmente lunga necessaria per ottenere l’AIC e evitare che la protezione venga persa prima ancora dell’inizio della commercializzazione del medicinale.
I testi applicabili?
L’esistenza del CCP è prevista da:
- il Regolamento n°1768/92 del Consiglio del 18 giugno 1992:
- per i CCP rilasciati dopo il 2 gennaio 1993 (articolo 23 del regolamento) se il brevetto riguarda un medicinale (articolo 2 del regolamento), il suo procedimento di ottenimento o il suo utilizzo (articolo 2 del regolamento insieme all’articolo 1.c);
- il Regolamento (CE) n° 1610/96 del Parlamento europeo e del Consiglio del 23 luglio 1996:
- per i CCP rilasciati dopo l’8 febbraio 1997 (articolo 23 del regolamento) se il brevetto riguarda un prodotto fitosanitario (articolo 2 del regolamento);
- gli articoli L611-2 CPI e L611-3 CPI:
- per i CCP rilasciati prima del 2 gennaio 1993.
Tanto per dire che le disposizioni del CPI non sono più molto utilizzabili oggi…
Titolo autonomo
Un CCP è un titolo autonomo, non prolunga la durata di un brevetto (anche se l’esistenza del brevetto è un prerequisito): conferisce una protezione propria e collegata a tale CCP.
Condizione per il rilascio dei CCP
Per ottenere un CCP, è necessario:
- richiederlo
- avere un brevetto in vigore (e non una domanda) che copra uno degli elementi precedentemente menzionati in base al testo applicabile;
- articolo 3.a del Regolamento n°1768/92 del Consiglio del 18 giugno 1992 o
- articolo 3.a del Regolamento (CE) n° 1610/96 del Parlamento europeo e del Consiglio del 23 luglio 1996;
- avere un’AIC in vigore che copra una specialità di tale medicinale (ossia il nome commerciale di un medicinale) coperto dal brevetto o se il medicinale o il prodotto fitosanitario brevettato ha ottenuto un’AIC
- articolo 3.b del Regolamento n°1768/92 del Consiglio del 18 giugno 1992 o
- articolo 3.b del Regolamento (CE) n° 1610/96 del Parlamento europeo e del Consiglio del 23 luglio 1996);
- non aver già richiesto un CCP (
- articolo 3.c del Regolamento n°1768/92 del Consiglio del 18 giugno 1992 o
- articolo 3.c del Regolamento (CE) n° 1610/96 del Parlamento europeo e del Consiglio del 23 luglio 1996);
- che tale AIC sia la prima nella Comunità
-
(
- articolo 3.d del Regolamento n°1768/92 del Consiglio del 18 giugno 1992 o
- articolo 3.d del Regolamento (CE) n° 1610/96 del Parlamento europeo e del Consiglio del 23 luglio 1996);
Ambito della protezione
La protezione conferita dal CCP è limitata alla specialità coperta dall’AIC (L611-3 CPI o articolo 4 del Regolamento n°1768/92 del Consiglio del 18 giugno 1992 o l’articolo 4 del Regolamento (CE) n° 1610/96 del Parlamento europeo e del Consiglio del 23 luglio 1996).
Inoltre, non è possibile ottenere un CCP più ampio (CGUE C-518/10 del 25 novembre 2011) o più ristretto (CGUE C-322/10 del 24 novembre 2011) rispetto alla rivendicazione del brevetto interessato (corrispondenza esatta): se la rivendicazione riguarda A1+A2, non è possibile ottenere un CCP su A1+A2+A3 (anche se l’AIC fosse concessa per A1+A2+A3) o su A1 da solo.
Durata della protezione
Principio
Il CCP consente di ottenere una protezione di durata non superiore (articolo 13 del Regolamento n°1768/92 del Consiglio del 18 giugno 1992 o l’articolo 13 del Regolamento (CE) n° 1610/96 del Parlamento europeo e del Consiglio del 23 luglio 1996):
- 5 anni a decorrere dalla scadenza del brevetto;
- e la differenza di tempo tra la data della prima AIC e la data di deposito del brevetto dalla quale è sottratto 5 anni.
In sintesi, la durata della protezione concessa dal CCP è:
- se l’AIC è concessa più di 10 anni dopo il deposito della domanda di brevetto: 5 anni;
- se l’AIC è concessa prima di 10 anni, la durata di 5 anni è ridotta di altrettanto.
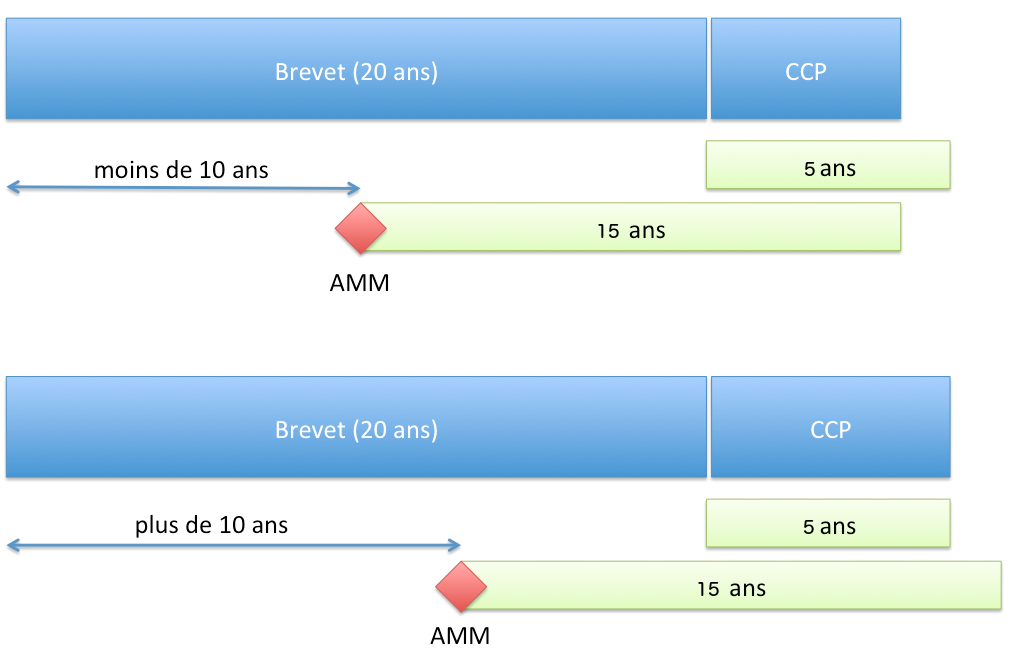 Durata della protezione conferita da un CCP (Regolamento UE)
Durata della protezione conferita da un CCP (Regolamento UE)Inizio del termine
La questione che si è posta è capire a cosa facesse riferimento la « data della prima autorizzazione all’immissione in commercio » :
- la data della decisione che concede l’AIC o
- la data della notifica di tale decisione o
- la data di rilascio.
Ciò può avere la sua importanza…
La CGUE ha avuto occasione di pronunciarsi sul tema (CGUE C‑471/14, 6 ottobre 2015) e ha indicato che la « data della prima autorizzazione all’immissione in commercio » era la data della notifica di tale decisione.
Durata negativa ?
A causa del metodo di calcolo precedentemente menzionato, è del tutto possibile che la durata di un CCP sia negativa.
Anche se di solito il titolare del brevetto non è interessato a ottenere un CCP di durata negativa, può eccezionalmente desiderarlo, in particolare se intende beneficiare di una proroga pediatrica menzionata di seguito.
La CGUE ha indicato che ciò era del tutto possibile (CGUE C‑125/10, 8 dicembre 2011).
Proroga pediatrica
Se il titolare di un CCP fornisce i risultati degli studi condotti secondo un piano di indagine pediatrica (in parte o interamente sulla fascia 0-18 anni) approvato nella sua domanda di AIC o di AIC complementare, ha diritto a una proroga di 6 mesi del suo CCP (Regolamento CE n. 1901/2006 del Parlamento Europeo e del Consiglio del 12 dicembre 2006, art 36.1).
Tuttavia, tale proroga non è possibile :
- se il medicinale è designato nell’AIC come destinato a una malattia rara pediatrica (è già concessa una protezione supplementare dell’AIC, vedi supra) (Regolamento CE n. 1901/2006 del Parlamento Europeo e del Consiglio del 12 dicembre 2006, art 36.4) ;
- se l’AIC beneficia di una protezione supplementare di un anno concessa in caso di nuova indicazione terapeutica (vedi supra) (Regolamento CE n. 1901/2006 del Parlamento Europeo e del Consiglio del 12 dicembre 2006, art 36.5).
Dans quel délai peut-on demander le CCP ? merci d’avance