



La procédure AMM : « Autorisation de Mise sur le Marché »
Qui est concerné par l’AMM ?
Les médicaments, avant d’être introduits sur le marché européen ou français, doivent recevoir une AMM (L5121-8 code de la santé publique).
Un médicament est (L 5111-1 du Code de la santé publique ou directive de la Communauté européenne 65/65/CEE du 26 janvier 1965, article 1er) :
- une substance ou composition présentée comme possédant des propriétés curatives ou préventives à l’égard de maladies, ou
- une substance ou composition pouvant être administré en vue d’établir un diagnostic médical, ou
- une substance ou composition pouvant être administré en vue de restaurer, de corriger ou de modifier des fonctions organiques.
Principales étapes de la conception d’un médicament
D’un point de vue général, voici les principales étapes de la vie d’un médicament, de la conception à la commercialisation.
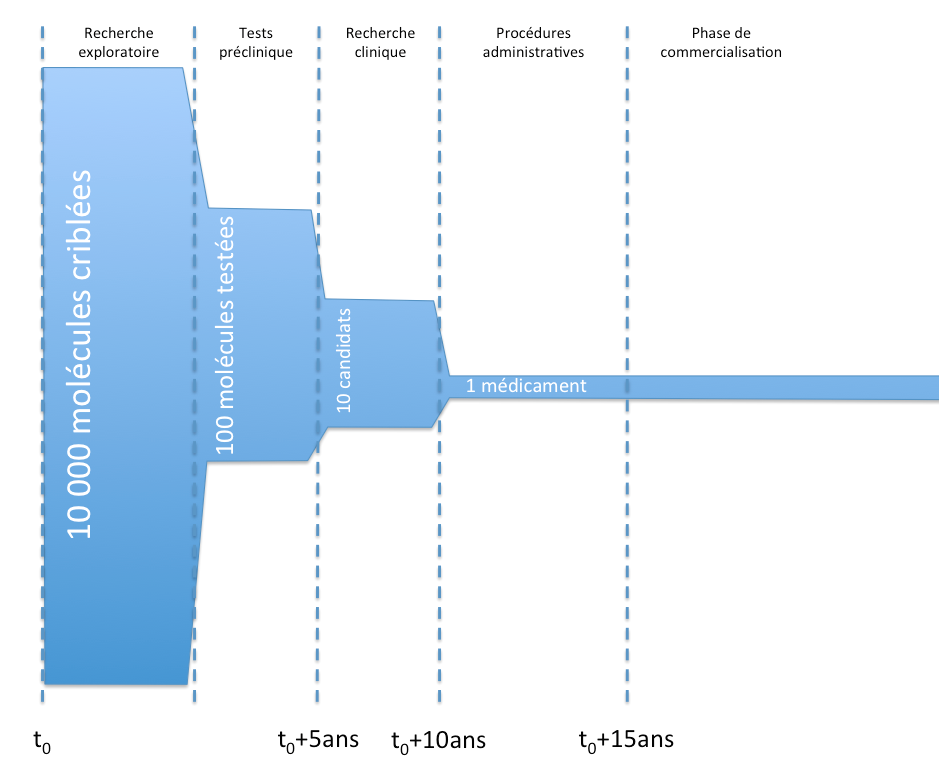 Vie d’un médicament et AMM
Vie d’un médicament et AMML’objet des essais précliniques est notamment de vérifier la tolérance des molécules in vitro chez l’animal, d’effectuer des tests toxicologiques et d’étudier la pharmacocinétique des molécules (en gros, de regarder à quelle vitesse les molécules agissent, sont absorbées, ou sont éliminées).
La recherche clinique se scinde en trois grandes phases :
- phase I :
- des essais chez des patients humains, sur une petite cohorte de volontaires sains.
- ces essais visent notamment à tester la tolérance chez les patients des molécules.
- phase II :
- des essais chez des patients humains, sur une cohorte de plus grande importance de volontaires malades (mais sans autres pathologies).
- ces essais visent notamment à établir un rapport bénéfice/risque de la molécule et de définir les doses d’efficacité.
- phase III :
- des essais chez des patients humains, sur une cohorte de grande importance de volontaires malades en conditions réelles.
- ces essais visent notamment à vérifier l’efficacité (étude en « double aveugle« ) et la sécurité sur le long terme.
Durant la phase de commercialisation, un mécanisme de « pharmacovigilance » (L5121-22 du code de la santé publique et suivants) est mis en place : des effets secondaires potentiels sont recueillis auprès des professionnels de santé et peuvent mener à des études observationnelles.
Par ailleurs, des études interventionnelles (dites de phase IV) peuvent être menées notamment si l’AMM le demande en raison du faible nombre de patients durant les essais cliniques (L5121-8-1 code de la Santé publique).
Enfin, durant la phase de commercialisation, des études cliniques secondaires peuvent être menées à l’initiative de l’industriel afin de tester son médicament sur une autre pathologie, une nouvelle population, dans le cadre d’une nouvelle stratégie thérapeutique, etc. afin de faire autoriser une nouvelle indication.
Procédures et modalités
Différentes procédures possibles
Pour obtenir une AMM, il est possible de choisir différentes voies :
- procédure nationale :
- ne vaut bien entendu que pour un seul pays
- procédure centralisée :
- couvre 27 pays européens ;
- peut être obligatoire pour certains médicaments (ex. biotechnologies ou nouvelles substances dans certains domaines comme cancer ou VIH) ;
- procédure de reconnaissance mutuelle :
- une AMM obtenue dans un pays de référence peut être reconnue par les autres pays ;
- procédure décentralisée :
- proche de la procédure de reconnaissance mutuelle, mais aucune AMM n’a encore été obtenue ;
- les dossiers sont déposés dans tous les pays, mais étudiés que par un pays de référence.
Dossier
Le dépôt d’un dossier AMM se fait auprès des autorités compétentes (française, européennes, etc.) dans un format qui est heureusement harmonisé (format CTD).
Le dossier AMM comporte notamment des indications :
- administratives (cela dépend des autorités compétentes) ;
- sur la qualité (procédé de fabrication, matière première utilisée, stabilité du produit, etc.) ;
- sur les essais précliniques ;
- sur la recherche clinique ;
- sur les mentions devant figurer sur les conditionnements et la notice à l’usage des patients (en annexe).
Prise de décision
Sur cette base, les autorités compétentes peuvent accorder l’AMM sur la base des informations de qualité, d’efficacité du médicament et de sécurité : un rapport bénéfice/risque est alors établi en tenant compte des autres traitements disponibles (L5121-9 Code de la santé publique).
La décision est importante, car elle fixe la dénomination, la forme du médicament, le mode d’administration, le dosage, les indications thérapeutiques, le prescripteur, la durée du traitement… L’ensemble de ces éléments va fixer indirectement le prix et donc la rentabilité du médicament.
Protection des données de l’AMM
Principe
Le dossier d’AMM n’est pas public pendant une certaine période, dite période de « protection des données » .
Pendant la période de « protection des données » , les autorités réglementaires ne peuvent :
- ni divulguer le dossier
- ni autoriser une autre société de faire référence à une AMM d’un tiers pour obtenir une autorisation.
La durée de la période de « protection des données » est de 8 + 2 + 1 ans (Règlement CE n°726/2004, art. 14(11)) :
- 8 ans de protection est accordée, période pendant laquelle personne ne peut accéder ou faire référence au dossier AMM (R5121-28 code de la santé publique) ;
- 2 ans de protection supplémentaire, période pendant laquelle il est possible de faire référence au dossier AMM, mais pendant laquelle il n’est pas possible d’obtenir d’AMM sur cette base (L5121-10-1 code de la santé publique) ;
- 1 an de protection supplémentaire (par rapport au 2 ans précédent), si pendant la période des 8 ans une nouvelle indication thérapeutique est accordée et que celle-ci apporte un avantage important(L5121-10-1 code de la santé publique).
Fin de la protection du dossier AMM et existence d’un brevet
Si le dossier AMM est accessible et qu’un tiers obtient une nouvelle AMM sur cette base (fin de la période 8+2+1), cela ne signifie pas qu’il ne sera pas contrefacteur si un brevet existe et est en vigueur.
Il convient de bien distinguer ces deux problématiques.
Protection et extension de gamme
Si après l’obtention d’une AMM, une nouvelle forme thérapeutique ou un nouveau dosage est autorisé (via une étude clinique secondaire), cela ne signifie pas qu’une nouvelle AMM est accordée.
Ainsi, une extension de gamme ne permet pas d’étendre la durée de protection d’une AMM (R5121-41-1 code de la santé publique ou Directive 2001/83/CE du Parlement Européen et du Conseil du 6 novembre 2001, art 6.1).
Protection et maladie orpheline
Une maladie orpheline ne touche pas plus de 5 personnes sur 10 000 dans la Communauté (ou que la commercialisation d’un médicament ne génère pas des bénéfices suffisants pour justifier un investissement) et qu’il n’existe pas encore de médicaments ayant été autorisés (tout du moins, avec une efficacité comparable) (Règlement n°141/2000 du Parlement européen et du Conseil, du 16 décembre 1999, article 3(1)).
Si une AMM est demandée pour une maladie orpheline, alors il existe une garantie qu’aucune autre AMM (même une extension d’AMM) ne sera accordée pour un médicament similaire (Règlement n°141/2000 du Parlement européen et du Conseil, du 16 décembre 1999, article 8.1) pendant 10 ans.
Ce délai de 10 ans peut être ramené à 6 ans, s’il établit avant la fin de la 5e année que la maladie n’est plus une maladie orpheline comme définie précédemment (Règlement n°141/2000 du Parlement européen et du Conseil, du 16 décembre 1999, article 8.2).
Néanmoins, cette exclusivité de 10 ans peut être réduite si (Règlement n°141/2000 du Parlement européen et du Conseil, du 16 décembre 1999, article 8.2) :
- le titulaire donne son accord ;
- le titulaire ne peut produire son médicament en quantité suffisante ;
- un nouveau demandeur démontre que son médicament est plus sûr, plus efficace ou cliniquement supérieur.
Protection et maladie orpheline pédiatrique
Le délai de 10 ans mentionné pour les maladies orphelines dans la section précédente est porté à 12 ans si la demande d’AMM comprend des résultats de l’ensemble des études réalisées selon un plan d’investigation pédiatrique approuvé (Règlement CE n°1901/2006 du Parlement Européen et du Conseil du 12 décembre 2006, art 37).
Il faut noter que si un CCP existe, il existe des dispositions particulières concernant les études pédiatriques (voir ci-dessous).
Fixation des prix et remboursement
Fixation des prix
La fixation du prix du médicament est fixée lors d’une négociation entre les industriels et le Comité Economique des Produits de Santé (ou CEPS, à défaut d’accord, le CEPS fixe unilatéralement le prix). Le prix, fixé par arrêté ministériel (L5123-1 code de la santé publique), tient compte :
- des prix des autres médicaments de même visée thérapeutique ;
- des volumes de ventes prévues ;
- des conditions d’utilisation ;
- du progrès par rapport aux traitements existants (ou AMSR, amélioration du service médical rendu).
Remboursement
En fonction du service médical rendu (SMR), il est décidé si le médicament est, en totalité ou en partie, pris en charge par la sécurité sociale. Les autres médicaments ne sont pas pris en compte dans cette évaluation.
La décision finale d’inscription au remboursement relève de la compétence des ministres chargés de la Santé et de la Sécurité sociale (L162-17-2-1 code de la sécurité sociale).
Le SMR peut être :
- important : remboursement de 65 % à 100 % (le taux maximal est accordé aux médicaments irremplaçables et coûteux) ;
- modéré : remboursement à 30 % ;
- faible : remboursement à 15 % ;
- insuffisant : non remboursé.
Génériques et bio-similaire
Définition
Un générique est un médicament ayant la même composition qualitative et quantitative en substance active et avec la même forme pharmaceutique qu’un médicament de référence (L5121-1 code de la santé publique, 5° a ou Directive 2001/83/CE du Parlement Européen et du Conseil du 6 novembre 2001, art 10.2 b).
Il faut, de plus, que la bioéquivalence entre le générique et le médicament de référence ait été prouvée.
Procédure simplifiée pour l’AMM
La demande d’AMM pour un générique est simplifiée.
Ainsi, le demandeur n’est pas tenu de fournir des résultats :
- des essais pharmacologiques et toxicologiques ;
- des essais cliniques.
Seules sont requises les données pharmaceutiques correspondant à la qualité des matières premières et au processus de fabrication.
Par ailleurs, il faut démontrer que le générique est similaire à un médicament ayant obtenus une AMM depuis au moins 10 ans (Directive 2001/83/CE du Parlement Européen et du Conseil du 6 novembre 2001, art 10.1) via une étude de bioéquivalence (i.e comportement équivalent dans l’organisme : absorption, distribution métabolisme et élimination).
Différences possibles
Il est donc possible d’avoir une présentation différente (ex. sirop vs. pilule) ou des excipients différents.
Il est également possible d’avoir une indication thérapeutique différente (i.e. liste des pathologies pour lequel le médicament peut être utilisé, Conseil d’État, 23 juillet 2003, n°246716).
Détermination du prix
Le prix est déterminé comme pour un médicament standard, mais celui-ci sera plus faible que le médicament de référence, car les coûts de recherche sont plus faibles.
Les Certificats Complémentaires de Protection ou CCP
Principe
Les CCP ont été crées afin pour tenir compte de la durée particulièrement longue nécessaire à l’obtention de l’AMM et éviter que la protection soit perdue avant même le début de la commercialisation du médicament.
Les textes applicables ?
L’existence du CCP est prévue par :
- le Règlement n°1768/92 du Conseil du 18 juin 1992 :
- pour les CCP délivrés après le 2 janvier 1993 (article 23 du règlement) si le brevet porte sur un médicament (article 2 du règlement), son procédé d’obtention ou son utilisation (article 2 du règlement ensemble article 1.c) ;
- le Règlement (CE) n° 1610/96 du Parlement européen et du Conseil du 23 juillet 1996 :
- pour les CCP délivrés après le 8 février 1997 (article 23 du règlement) si le brevet porte sur un produit phytosanitaire (article 2 du règlement) ;
- les articles L611-2 CPI et L611-3 CPI :
- pour les CCP délivrés avant le 2 janvier 1993.
Autant dire que les dispositions du CPI ne sont plus trop utilisables aujourd’hui…
Titre autonome
Un CCP est un titre autonome, il ne prolonge pas la durée d’un brevet (même si l’existence du brevet est un prérequis) : il confère une protection propre et attachée à ce CCP.
Condition d’octroi des CCP
Pour obtenir un CCP, il faut :
- le demander
- article 7 du Règlement n°1768/92 du Conseil du 18 juin 1992 ou
- article 7 du Règlement (CE) n° 1610/96 du Parlement européen et du Conseil du 23 juillet 1996 ;
- avoir un brevet en vigueur (et non une demande) couvrant un des éléments mentionnés précédemment en fonction du texte applicable ;
- article 3.a du Règlement n°1768/92 du Conseil du 18 juin 1992 ou
- article 3.a du Règlement (CE) n° 1610/96 du Parlement européen et du Conseil du 23 juillet 1996 ;
- avoir une AMM en vigueur couvrant une spécialité de ce médicament (i.e. le nom de marque commerciale d’un médicament) couvert par le brevet ou si le médicament ou le produit phytosanitaire breveté a fait l’objet d’une AMM
- article 3.b du Règlement n°1768/92 du Conseil du 18 juin 1992 ou
- article 3.b du Règlement (CE) n° 1610/96 du Parlement européen et du Conseil du 23 juillet 1996) ;
- ne pas avoir déjà demander un CCP
- article 3.c du Règlement n°1768/92 du Conseil du 18 juin 1992 ou
- article 3.c du Règlement (CE) n° 1610/96 du Parlement européen et du Conseil du 23 juillet 1996) ;
- que cette AMM soit la première dans la Communauté
- article 3.d du Règlement n°1768/92 du Conseil du 18 juin 1992 ou
- article 3.d du Règlement (CE) n° 1610/96 du Parlement européen et du Conseil du 23 juillet 1996) ;
Portée de la protection
La protection conférée par le CCP est limitée à la spécialité couverte par l’AMM (L611-3 CPI ou article 4 du Règlement n°1768/92 du Conseil du 18 juin 1992 ou l’article 4 du Règlement (CE) n° 1610/96 du Parlement européen et du Conseil du 23 juillet 1996).
Par ailleurs, il n’est pas possible d’obtenir un CCP plus large (CJUE C-518/10 du 25 novembre 2011) ou plus restreint (CJUE C-322/10 du 24 novembre 2011) que la revendication du brevet visé (exacte concordance) : si la revendication vise A1+A2, il n’est pas possible d’avoir un CCP sur A1+A2+A3 (quand bien même l’AMM serait accordée pour A2+A2+A3) ou sur A1 seul.
Durée de protection
Principe
Le CCP permet d’obtenir une protection d’une durée ne pouvant dépasser (article 13 du Règlement n°1768/92 du Conseil du 18 juin 1992 ou l’article 13 du Règlement (CE) n° 1610/96 du Parlement européen et du Conseil du 23 juillet 1996) :
- 5 ans à compter de l’expiration du brevet ;
- et la différence de temps entre la date de la première AMM et la date de dépôt du brevet auquel est soustrait 5 ans.
Pour résumer, la durée de protection accordée par le CCP est de :
- si l’AMM est accordée plus de 10 ans après le dépôt de la demande de brevet : 5 ans ;
- si l’AMM est accordée plus rapidement que 10 ans, la durée de 5 ans est réduite d’autant.
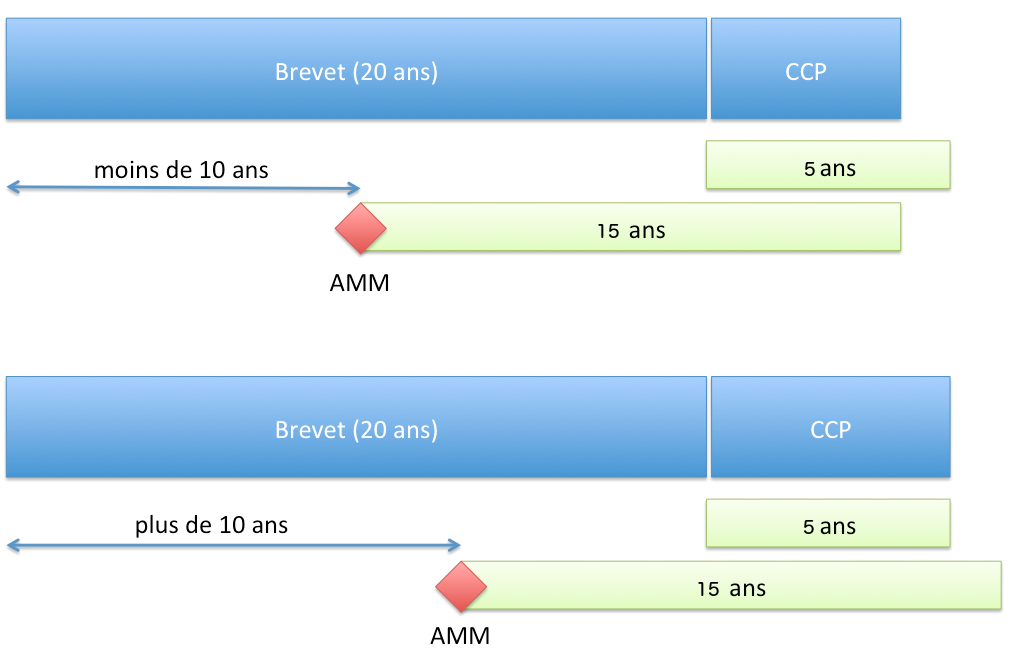 Durée de protection conférée par un CCP (Règlement EU)
Durée de protection conférée par un CCP (Règlement EU)Point de départ du délai
La question qui s’est posée est de savoir si à quoi faisait référence la « date de la première autorisation de mise sur le marché » :
- la date de la décision accordant l’AMM ou
- la date de la notification de cette décision ou
- la date d’octroi.
Cela peut avoir son importance…
La CJUE a eu l’occasion de se prononcer sur le sujet (CJUE C‑471/14, 6 octobre 2015) et a indiqué que la « date de la première autorisation de mise sur le marché » était la date de la notification de cette décision.
Durée négative ?
Du fait de la méthode de calcul précédemment mentionnée, il est tout à fait possible que la durée d’un CCP soit négative.
Même si habituellement le titulaire du brevet n’est pas intéressé par le fait d’obtenir un CCP de durée négatif, il peut exceptionnellement le désirer, notamment s’il compte bénéficier d’une prolongation pédiatrique mentionnée ci-dessous.
La CJUE a indiqué que cela était tout à fait possible (CJUE C‑125/10, 8 décembre 2011).
Prolongation pédiatrique
Si le titulaire d’un CCP fournit les résultats des études réalisées selon un plan d’investigation pédiatrique (en partie ou en totalité sur la tranche 0 à 18 ans) approuvé dans son dépôt d’AMM ou d’AMM complémentaire, celui-ci a le droit à une prolongation de 6 mois de son CCP (Règlement CE n°1901/2006 du Parlement Européen et du Conseil du 12 décembre 2006, art 36.1).
Néanmoins, cette prolongation n’est pas possible :
- si le médicament est désigné dans l’AMM comme visant une maladie orpheline pédiatrique (une protection supplémentaire de l’AMM est déjà accordée, voir supra) (Règlement CE n°1901/2006 du Parlement Européen et du Conseil du 12 décembre 2006, art 36.4) ;
- si l’AMM bénéficie d’une protection supplémentaire d’un an accordée en cas de nouvelle indication thérapeutique (voir supra) (Règlement CE n°1901/2006 du Parlement Européen et du Conseil du 12 décembre 2006, art 36.5).
Dans quel délai peut-on demander le CCP ? merci d’avance